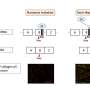
Figure 1. Genotype-phenotype correlation for renal survival in Alport syndrome. Credit: Kobe University
A large-scale analysis of the clinical characteristics of Alport syndrome in Japanese patients has revealed that the effectiveness of existing treatment with ACE inhibitors and/or angiotensin receptor blockers (RAS inhibitors) varies depending on the type of mutation in the syndrome's causal gene (COL4A5). RAS inhibitors are widely administered to patients with chronic kidney diseases as they are known to preserve kidney functions, and they are also effective against Alport Syndrome. In addition to providing proof of the effectiveness of RAS inhibitor treatment in Japanese patients with Alport syndrome, the researchers revealed for the first time in the world that the degree of effectiveness depends on the genotype.
The research group included Assistant Professor Yamamura Tomohiko, Professor Iijima Kazumoto and Professor Nozu Kandai et al. of the Department of Pediatrics, Kobe University Graduate School of Medicine.
These results were published online in the journal Kidney International on July 23, 2020.
Main Points
- Alport syndrome is a hereditary disorder characterized by nephritis, hearing loss and eye abnormalities (such as anterior lenticonus or cataracts).
- X-linked Alport syndrome is the most common form and men in particular exhibit severe symptoms. Those who have a severe mutation (such as a nonsense mutation) in Alport syndrome's casual gene COL4A5 are known to develop end-stage kidney disease (ESKD) in their early twenties.
- One promising treatment involves using ACE inhibitors and/or angiotensin receptor blockers (RAS inhibitors) to preserve kidney functions. So far, studies conducted outside Japan have shown these treatments to be effective for lowering urinary protein levels and suppressing the progression of kidney function deterioration.
- This research team conducted a large scale survey of Japanese male patients with Alport syndrome. They compared the median results of a group of patients who were prescribed RAS inhibitors with a group of patients who didn't take the drug. They found that the age at which patients taking RAS inhibitors progressed to ESKD was delayed by over 20 years, demonstrating the effectiveness of this treatment.
- The team also compared the effectiveness of the treatment for those with minor mutations (such as missense mutations) and those with severe mutations in the casual gene. Although RAS inhibitors were effective in treating those with severe mutations, they were comparatively more effective in those with minor mutations.
- For the first time in the world, this research demonstrated that the effectiveness of RAS inhibitors differs depending on the genotype of Alport syndrome.

Figure 2. Renal survival proportion based on the presence of RAS inhibitor treatment. Credit: Kobe University
Alport syndrome (AS) is the second most commonly occurring hereditary kidney disease after autosomal dominant polycystic kidney disease (ADPKD). There is one case of Alport syndrome in every 5,000 to 10,000 people. It is characterized by hearing loss, eye abnormalities and kidney disease, often progressing to end stage kidney disease (ESKD). Alport syndrome is divided into three groups according to how it is inherited; X-linked AS, autosomal recessive AS and autosomal dominant AS with approximately 80% of cases being X-linked Alport syndrome (XLAS).
XLAS develops due to pathological mutations in the COL4A5 gene that encodes the type IV collagen α5 chain. In particular, male patients with XLAS are likely to have severe symptoms and around 90% of them experience ESKD by the age of 40. This requires them to undergo renal replacement therapies such as dialysis and kidney transplants. However, there is still no specialized treatment for Alport syndrome itself.
Men with severe types of Alport syndrome (such as those caused by nonsense mutations in the COL4A5 gene) develop ESKD over ten years earlier than those with milder forms (such as those caused by missense mutations). A possible treatment for Alport syndrome involves using ACE inhibitors and/or angiotensin receptor blockers (RAS inhibitors) to preserve kidney functions. So far, studies conducted outside Japan have shown these treatments to be effective for lowering urinary protein levels and suppressing the progression of kidney function deterioration.
Up until now, this research team has established a comprehensive genetic diagnosis system for Alport syndrome, which has enabled them to conduct genetic diagnoses of Japanese patients. This time, the researchers conducted a retrospective investigation into the clinical characteristics of the disorder in 430 male patients with XLAS. From these results and by drawing on the findings of other research, they revealed the following:

Figure 3. Renal survival proportion based on the presence of genotype-dependent RAS inhibitor treatment. Credit: Kobe University
- It was possible to analyze the clinical data of 422 of these patients, and the results showed that the median age for progression to ESKD was 35.
- There was a very strong correlational relationship between genotype and the median age for progression to ESKD. The median age was 18 for those with nonsense mutations (Figure 1: black survival curve), whereas it was 40 in those with missense mutations (Figure 1: yellow survival curve). This is a difference of 22 years (Figure 1).
- The data also revealed a connection between the symptom of hearing loss and the median age at which ESKD developed. The median age in patients with hearing loss was 28, in those without hearing loss it was 55. This showed that renal symptoms were more severe in cases with hearing loss.
- Clinical data from 207 patients that showed whether or not they were prescribed RAS inhibitors was also analyzed. The results revealed that those who didn't receive RAS inhibitor treatment developed ESKD by a median age of 28, whereas over half of those prescribed with RAS inhibitors did not develop ESKD before the age of 50. In other words, it was shown that this drug could delay the onset of ESKD by over 20 years (Figure 2).
- Subsequently, the researchers evaluated the effectiveness of RAS inhibitors depending on whether the patient had a severe mutation or a minor mutation. In patients with minor mutations, those who were not administered RAS inhibitors developed ESKD at a median age of 33 (Figure 3: red survival curve), whereas over half of those who received treatment with the drug did not develop ESKD before a median age of 50 (Figure 3: blue survival curve).
On the other hand, in patients with severe mutations, the group who weren't prescribed RAS inhibitors progressed to ESKD by a median of 16 years of age (Figure 3: black survival curve), whereas those receiving RAS inhibitor treatment developed ESKD at a median age of 28 (Figure 3: green survival curve). Therefore, the drug was effective in treating Alport syndrome caused by severe mutations as it delayed the onset of ESKD in patients by a median of 12 years. However the treatment was shown to be less effective than in patients with minor mutations (Figure 3).
RAS inhibitors were effective, even for patients with severe mutations, however the median age for progression to end stage renal failure for those with severe forms of the syndrome was 28. The current team is currently developing a treatment method (Exon skipping therapy) for patients with severe mutations, and the above results are likely to increase the demand for this treatment's development.
More information: Tomohiko Yamamura et al. Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked Alport syndrome., Kidney International (2020). DOI: 10.1016/j.kint.2020.06.038
Journal information: Kidney International
Provided by Kobe University