Credit: CC0 Public Domain
A research team led by Prof. Zhang Shihua from the Academy of Mathematics and Systems Science of the Chinese Academy of Sciences has proposed a new computational tool, STAGATE, to decipher tissue substructures from spatial resolved transcriptomics (STs).
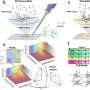
The model uses artificial intelligence technology to integrate spatial location information and gene expression profile of spatial spots. In this algorithm, a graph attention autoencoder is introduced, with a graph attention mechanism in the middle hidden layer, which can learn the heterogeneous similarities between neighboring spots adaptively. Results were published in Nature Communications.
Deciphering tissue substructures or spatial domains (i.e., tissue regions with similar spatial expression patterns) is one of the great challenges presented by STs. For example, the laminar organization of the human cerebral cortex is especially related to its biological functions, in which cells residing within different cortical layers often differ in expressions, morphology and physiology. However, most existing clustering methods do not efficiently use the available spatial information, which results in discrete tissue substructures. Also, they are highly susceptible to technical noise.
According to the researchers, the new model converts the spatial location information into a spatial neighbor network between spatial spots, and then feeds the gene expression information and the spatial network into a graph attention autoencoder to learn a low-dimensional representation of spot.
In addition, it combines the characteristics of 10x Visium data, and suggests a cell type-aware module based on expression information pre-clustering to better describe the boundary of the cell space domain.
Intriguingly, the new model can reduce the batch effect between different sections by introducing a spatial network between adjacent sections, and improve the performance of three-dimensional tissue sub-structures.
The superiority of STAGATE for deciphering tissue substructures or spatial domains has been validated in diverse datasets. It is worth noting that it can be used to analyze spatial transcriptomics data of different sequencing technology platforms (including 10x Visium, Slide-seq, Stereo-seq, etc.) with diverse space resolutions.
"With the rapid development of spatial omics technology and the continuous accumulation of data, this new model STAGATE can facilitate the precise analysis of large-scale spatial transcriptome data and advance our understanding of tissue substructures," said Zhang Shihua, an expert of machine learning and computational biology, and lead author of the study.
More information: Kangning Dong et al, Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder, Nature Communications (2022). DOI: 10.1038/s41467-022-29439-6
Journal information: Nature Communications
Provided by Chinese Academy of Sciences