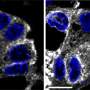
is shown overlapping with the Golgi apparatus (red), which indicates that phosphotransferase is located in the Golgi, where it should be. Credit: Eline Van Meel, PhD")
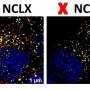
In normal cells, phosphotransferase (green) is shown overlapping with the Golgi apparatus (red), which indicates that phosphotransferase is located in the Golgi, where it should be. Credit: Eline Van Meel, PhD
(Medical Xpress)—To remain healthy, the body's cells must properly manage their waste recycling centers. Problems with these compartments, known as lysosomes, lead to a number of debilitating and sometimes lethal conditions.
Reporting in the Proceedings of the National Academy of Sciences (PNAS), researchers at Washington University School of Medicine in St. Louis have identified an unusual cause of the lysosomal storage disorder called mucolipidosis III, at least in a subset of patients. This rare disorder causes skeletal and heart abnormalities and can result in a shortened lifespan. But unlike most genetic diseases that involve dysfunctional or missing proteins, the culprit is a normal protein that ends up in the wrong place.
"There is a lot of interest and study about how cells distribute proteins to the right parts of the cell," said senior author Stuart A. Kornfeld, MD, PhD, the David C. and Betty Farrell Professor of Medicine. "Our study has identified one of the few examples of a genetic disease caused by the misplacement of a protein. The protein functions just fine. It just doesn't stay in the right place."
The right place, in this case, is the Golgi apparatus, the cell's protein packaging center. The protein in question – phosphotransferase – normally resides in the Golgi, where its job is to attach address labels to proteins bound for the lysosome. There are 60 such lysosomal proteins, and all of them must be properly labeled if they are to end up in a lysosome, where they recycle waste.
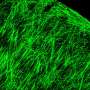
 is spread beyond the Golgi (red). Outside the Golgi, this wayward phosphotransferase is no longer able to perform its job of properly addressing enzymes bound for the lysosome. Credit: Eline Van Meel, PhD")
In mutant cells, the protein phosphotransferase (green) is spread beyond the Golgi (red). Outside the Golgi, this wayward phosphotransferase is no longer able to perform its job of properly addressing enzymes bound for the lysosome. Credit: Eline Van Meel, PhD
Kornfeld and his colleagues, including first author Eline van Meel, PhD, postdoctoral research associate, showed that the phosphotransferase protein responsible for adding the address label starts out in the Golgi as it should, but seems to lack the signal to keep it there.
"Under normal circumstances, the phosphotransferase moves up through the Golgi, but then it's recaptured and sent back," Kornfeld said. "Our study shows that the mutant phosphotransferase moves up but is not recaptured. Ironically, the phosphotransferase that escapes the Golgi ends up in the lysosomes, where it is degraded."
Because phosphotransferase gradually wanders away from the Golgi, a low level of lysosomal enzymes end up being properly addressed, but at perhaps 20 percent of the normal amount.
"In many lysosomal storage disorders, such as Tay-Sachs or Gaucher's disease, only one out of the 60 enzymes is missing from the lysosome," Kornfeld said. "But the mislocalization of phosphotranferase causes the misdirection of all 60 lysosomal enzymes."
While the errant phosphotransferase ends up being degraded in the lysosome, the resulting misdirected lysosomal proteins end up in the bloodstream. As a result, children with this disorder have lysosomal proteins in their blood at levels 10 to 20 times higher than normal. But because some get to the lysosome at a low level, people with mucolipidosis III don't have the most severe form of the disease.
"Type III patients live into adulthood, but they're very impaired," said Kornfeld. "They have joint and heart problems and have trouble walking. In the most severe form, type II, there is zero activity of phosphotransferase. None of the 60 enzymes are properly tagged, so these patients' lysosomes are empty. Children with type II usually die by age 10."
Having implicated wayward phosphotransferase in this lysosomal storage disorder, Kornfeld and his colleagues are investigating what goes wrong that allows it to escape the Golgi.
"We think there must be some protein in the cell that recognizes phosphotransferase when it gets to the end of the Golgi, binds it and takes it back," said Kornfeld. "Now we're trying to understand how that works."
More information: Van Meel, E, Qian Y, Kornfeld SA." Mislocalization of phosphotransferase as a cause of mucolipidosis III ab." PNAS. Online Feb. 18, 2014.
Journal information: Proceedings of the National Academy of Sciences