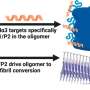
From left to right, the experts Susana Balcells, Daniel Grinberg and Roser Urreizti at the Faculty of Biology of the University of Barcelona
Opitz C syndrome is a genetic disease that causes severe disabilities in patients and has been diagnosed in three people in the Iberian Peninsula, and 60 people in the world. A team led by the professors Daniel Grinberg and Susana Balcells, from the University of Barcelona and the Biomedical Research Networking Center of Rare Diseases (CIBERER) has now identified a gene that causes Opitz C syndrome in the only patient in Catalonia diagnosed with this severe congenital disease. This new scientific advance is a first step to discovering the genetic bases of this syndrome which, so far, has no treatment, prenatal diagnosis or genetic counseling.
The new study, published in the journal Scientific Reports, has the participation of John M. Opitz (University of Utah, United States), Giovanni Neri (Catholic University of the Sacred Heart, Italy) and a wide group of experts of the Center for Genomic Regulation (CRG) and the Department of Clinical and Molecular Genetics of the University Hospital Vall d'Hebron (VHIR).
Opitz C syndrome: rare but not invisible
The genetic bases of this ultra-minority disease, described for the first time in 1969 by John M. Opitz, are still unknown. It is generally thought that its origin is caused by the apparition of dominant -maternally silenced- novo mutations. At the moment, the diagnose is clinical and it is based on the symptomatology presented on patients with different degrees (trigonocephaly, learning disability, psychomotor disability, etc.) and which, in lots of cases, coincides with similar minority pathologies such as the syndromes of Schaaf-Yang, Bohring-Opitz and Prader-Willi.
In the new study, the experts described for the first time, the existence of a novo mutation –p.Q638— located in the gene MAGEL2 of the only diagnosed person with Opitz C syndrome in Catalonia. Identifying this mutation, found in the Prader-Willi Region on chromosome 15, widens the knowledge horizons on genetics and the possibilities for a diagnosis on these rare diseases.
"The p.Q638* mutation, identified in the gene MAGEL2, coincides with the one described concurrently and independently in a patient with Schaaf-Yang syndrome, a new minoritary disease affecting fifty people in the world. The first cases were described on a scientific bibliography in 2013 by the team of Professor Christian Schaaf, from the Baylor College of Medicine, Houston," says Professor Daniel Grinberg, member of the Institute of Biomedicine of the University of Barcelona (IBUB), the Research Institute of Sant Joan de Déu (IRSJD) and CIBERER.
"Consequently, from a genetic diagnosis perspective –says DanieL Grinberg- this patient initially diagnosed with Opitz C in Catalonia would correspond to the group of patients with Schaaf-Yang syndrome."
Genetics will define the limits of rare diseases
Identifying the genes that cause a disease is a breakpoint to understand the pathology and set new future therapeutic approaches that improve the quality of life of the patients. In the new study, the teams of the UB and the CRG applied techniques of DNA massive sequencing (exome and genome), a powerful methodology that allows identifying altered genes in each patient.
According to Susana Balcells, tenured lecturer at the UB and also member of IBUB and CIBERER, "what we can see from a clinical symptomatology view in these kinds of diseases which are so hard to study and diagnose, is far from the initial molecular defect that generates the disease."
"All these clinical doubts –continued Balcells- will be solved with genetics, which will define the limits of these rare diseases and will ease the scientific consensus on the diagnosis and genetic causes that create them."
According to Luis Serrano, director of CRG, "projects like this one show the important role of genomics in the future of medicine and the way on which we diagnose and treat diseases. To understand the diseases and offering not only a diagnosis but also approaches to possible treatments is very relevant in minority diseases. It is a satisfaction for the CRG to contribute with our knowledge and advanced technologies in a project that gives hope to a vulnerable collective," concluded the researcher.
More information: Roser Urreizti et al. A De Novo Nonsense Mutation in MAGEL2 in a Patient Initially Diagnosed as Opitz-C: Similarities Between Schaaf-Yang and Opitz-C Syndromes, Scientific Reports (2017). DOI: 10.1038/srep44138
Journal information: Scientific Reports
Provided by University of Barcelona