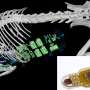
Age 2 months: normal echogenicity and clearly visible internal septa. (B) Age 14 months, 6 months after onset of disease: a pathologic increase in background echogenicity in comparison to subcutaneous fat tissue and less definable intramuscular septa. (C) Age 17 months, after eight months of nusinersen therapy: partial recovery with increase in volume and normalization of echogenicity in the rectus femoris. Credit: Wolfgang Müller-Felber, MD")
Sonographic findings of quadriceps in child diagnosed with SMA: (A) Age 2 months: normal echogenicity and clearly visible internal septa. (B) Age 14 months, 6 months after onset of disease: a pathologic increase in background echogenicity in comparison to subcutaneous fat tissue and less definable intramuscular septa. (C) Age 17 months, after eight months of nusinersen therapy: partial recovery with increase in volume and normalization of echogenicity in the rectus femoris. Credit: Wolfgang Müller-Felber, MD
The advent of therapeutic interventions for spinal muscular atrophy (SMA) has increased the importance of presymptomatic diagnosis and treatment. When to start treatment in children with less severe disease remains controversial. Now, in a report published in the Journal of Neuromuscular Diseases, German researchers argue for an earlier start of treatment to prevent permanent nerve damage, challenging recommendations originally proposed by a group of American experts that suggests a strict follow-up strategy for children expected to have less severe disease.
"Recent advances in the treatment of spinal muscular atrophy argue in favor of newborn screening (NBS) for SMA. While a highly sensitive and specific method exists to detect the most important causative mutation of spinal muscular atrophy, a homozygous deletion of the SMN1 gene, prognosis of the clinical course of the disease remains difficult. The most important disease modifier is the copy number of the SMN2 gene," explained lead investigator Wolfgang Müller-Felber, MD, Dr. V. Hauner Children's Hospital, Department of Pediatric Neurology and Developmental Medicine, LMU—University of Munich, Munich, Germany.
SMA is an autosomal recessive disease that is characterized by the degeneration of spinal cord alpha motor neurons. It is the most frequent genetic cause of death in infants. The disease is caused by a lack of survival motor neuron (SMN) protein, which leads to the loss of spinal motor neurons. The severity of disease ranges from type 0, the most serious congenital form, type 1, which is characerized by motor impairment beginning within the first six months, inability to sit, and death due to respiratory failure, to type 3 (symptoms manifest after walking begins although the ability to walk may be lost over time). Genetically, the disease is attributed to the presence of a homozygous deletion of the SMN1 gene while the number copies of the highly related SMN2 gene is known to modify disease, with four or more SMN2 copies suggesting milder disease progression.
The researchers reported their experiences with children identified through an NBS program for SMA that was conducted in Germany from January 2018 to November 2019. After screening nearly 280,000 newborns, 38 children were found to have biallelic loss of SMN1 confirming a genetic diagnosis of SMA. Of those, 15 (40%) had four or more SMN2 copies, prediciting a likely milder disease course. The children were treated according to the 2018 American SMA NBS Multidisciplinary Working Group recommendations, which include regular clinical, electrophysiological, and muscle ultrasound exams until the onset of SMA symptoms. According to the recommendations, treatment should begin only when symptoms appear.
"We found that inconsistent compliance, technical issues, and difficulties with the correct interpretation of clinical symptoms can make the use of the 2018 American SMA NBS Multidisciplinary Working Group recommendations problematic and thus may have a negative impact on the correct timing of treatment initiation," noted Prof. Dr. Müller-Felber. These recommendations have now been modified and are published in the same issue of the Journal of Neuromuscular Diseases.
One specific case in the German study concerned a child in whom the SMN2 copy number was intially incorrectly reported as high and treatment thus not initiated. The child then developed symptoms of SMA at eight months of age. After onset of symptoms, it took another five weeks until the insurance company approved treatment. During the delay, the child continued to deteriorate until he was unable to bear any weight on his legs. Once treatment began, the child partially recovered. "We assume early treatment prevents or limits damage to motor neurons, helps recovery, and prevents progression, thus arguing for the earliest possible treatment," commented Prof. Dr. Müller-Felber.
Another case in the German study revealed that two affected siblings were not correctly diagnosed as SMA patients before SMA was detected in a newborn sibling.
"In view of the results of our study, an inflexible follow-up-only strategy seems to be too risky for most children, even when four copies of the SMN2 gene make a benign course of the disease probable. NBS programs increase the chances for early diagnosis as well as discovering missed diagnoses. An earlier start of treatment should be considered in all children diagnosed by NBS in order to prevent irreversible damage of motor neurons," stated Prof. Dr. Müller-Felber.
More information: Wolfgang Müller-Felber et al, Infants Diagnosed with Spinal Muscular Atrophy and 4 SMN2 Copies through Newborn Screening – Opportunity or Burden?, Journal of Neuromuscular Diseases (2020). DOI: 10.3233/JND-200475
Provided by IOS Press