Treatment restores some function in animal models of spinal muscular atrophy
In work involving several new generations of mouse model development, Jackson Laboratory (JAX) researchers have tested a therapeutic intervention for spinal muscular atrophy (SMA) that restores some function lost due to a mutation in one gene (SMN1) and amplifies the levels of protective genes (SMN2).
Moreover, unlike current interventions, the therapy appears to work after symptoms of SMA have already appeared, and may not need to be administered directly into the central nervous system.
SMA is a neurodegenerative disorder caused by mutations that cause loss of function in the Survival Motor Neuron (SMN) 1 gene. SMN1, as its name implies, is vital for proper motor neuron function and maintenance. SMA patients have varying levels of severity because of the presence of a homolog gene, SMN2, in humans. SMN2 provides some protection because a small number of the proteins produced from it function like SMN1. As a result, SMA disease prognosis depends on how many copies of SMN2 are present. Two or fewer copies of SMN2 leads to earlier onset, more severe disease, while three or more copies provide enough function to result in a later onset, milder form of SMA.
SMA has proven to be difficult to model in mice. Current mouse models expressing small amounts of SMN protein die very early, by about two weeks of age. Yet, increasing this protein level by just a small amount renders the mice almost normal. This makes therapy testing very difficult and doesn't model the symptoms or processes associated with the less severe types of the disease.
A research group led by Cat Lutz, Ph.D., director of the JAX Rare and Orpan Disease Center, is working to improve the ability to model SMA in mice. As published on October 12 in the Proceedings of the National Academy of Sciences, their first step was to genetically engineer mouse models with increasing copy numbers of human SMN2. Through this work they were able to develop a mouse model with very mild neuromuscular defects that lives a near normal life span, and little motor neuron denervation. So a better model was still needed.
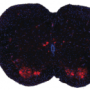
The researchers developed a model with SMN1, human SMN2 and a third allele (dubbed the C allele) that combines parts of mouse SMN with parts of human SMN2. This combination produced a mouse that had a relatively long life span compared to severe mutants (about 100 days) combined with SMA-like neuromuscular disease phenotype.
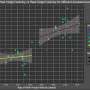
The new model, called "Burgheron" to honor the researchers who made the integrated alleles available, allowed Lutz and her colleagues to test a current therapeutic intervention that restores SMN levels and has a significant effect in severe models. The therapy, known as an antisense oligonucleotide, worked to restore SMN levels in the Burgheron model too, and even injections 25 days after symptoms had begun increased life span and restored motor unit function in the muscles.
"The majority of the preclinical data that we have to date comes from type I animal models and suggests that early intervention in the treatment of SMA is necessary," says Lutz. "However, we really don't have good preclinical data that supports whether this is also true for type II/III patients, who represent the majority of patients living with SMA. Our data indicate that post-symptomatic treatment, well into the disease state in mice, has a clinically relevant therapeutic benefit."
Human trials for therapeutics involving antisense oligonucleotides are delivered directly into the central nervous system (CNS). Peripheral application in the mouse model had benefits here, even at the neuromuscular junction, raising the question of what might be alternative delivery mechanisms in humans.
"The benefit may not be restricted to CNS delivery," says Lutz, "which opens up a whole new potential class of compounds and possibilities for combination therapies. Further study in humans will be needed, but the prospect of non-CNS delivery is of significant potential benefit for patients."
More information: Laurent P. Bogdanik et al. Systemic, postsymptomatic antisense oligonucleotide rescues motor unit maturation delay in a new mouse model for type II/III spinal muscular atrophy, Proceedings of the National Academy of Sciences (2015). DOI: 10.1073/pnas.1509758112