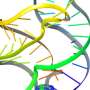
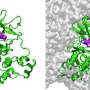
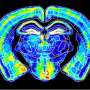
Computer simulation methods developed by Pratyush Tiwary’s lab identified two optimum pathways that Gleevec, a cancer drug, could take to unbind from the protein. The “fast” pathway allows Gleevec to leave the protein three times faster than that of the “slow” pathway, ultimately leading to drug resistance. Credit: Pratyush Tiwary
Imatinib, better known as Gleevec, was hailed as a "miracle" cancer drug when it entered the market in the early 2000s. Though it has been highly successful at treating early-stage chronic myeloid leukemia (CML)—a rare cancer that forms in bone marrow cells—many late-stage patients experience drug resistance caused by mutations of vital proteins in the body.
A new study led by Pratyush Tiwary, an assistant professor in the University of Maryland's Department of Chemistry and Biochemistry and Institute for Physical Science and Technology, used computational chemistry to figure out what causes resistance to Gleevec at the molecular level.
The study was published online in the journal Angewandte Chemie International Edition on April 29, 2022. The journal's editors labeled it a "Hot Paper"—a distinction granted to papers of utmost interest and importance. The metrics developed by Tiwary's lab could have broad applications for the pharmaceutical industry, potentially yielding drugs that target a variety of diseases with higher rates of efficacy.
"What this work really does is allow us to figure out which mutations will make any given drug ineffective—and why," Tiwary said. "Often these could not be predicted just by looking at the structure of a protein. You need these sophisticated simulations that my group does in order to look at this. In physical chemistry, this is known as the thermodynamics versus kinetics debate."
For this study, Tiwary and his co-authors focused on Gleevec, which controls cancer by blocking a particular protein known as a kinase. In some cases, the kinase may mutate in response to being blocked by the drug, in response to environmental factors or simply by chance.
"In some sense, it's similar to COVID, where mutation happens in the spike protein," Tiwary explained. "One little part of the protein will change into something else that makes the drug ineffective."
Mutations at the spot where Gleevec binds to a protein—known as a drug-binding pocket—can paint a clearer picture of why drug resistance occurs. However, Tiwary and his co-authors set out to answer a trickier question: Why does one particular mutation called N368S cause drug resistance, even though it doesn't appear to affect the way that the drug binds?
This is where a supercomputer comes in. Tiwary's lab used machine learning and a subdiscipline of chemistry known as statistical mechanics to design computer simulation methods capable of examining extremely small structures—think atoms and molecules—in extremely high resolution over the course of minutes, hours and even days. This is unique because, in most cases, supercomputers can only capture microseconds of data at such a high resolution.
The model works by alternating between rounds of machine learning to estimate a pathway that Gleevec could take to leave the protein. This information is used to perform new simulations, with each simulation round producing new data that trains the machine learning model to learn an improved pathway. The process continues until machine learning and simulations agree with each other.
The simulations ultimately determined two optimum pathways that describe how Gleevec unbinds from the protein and its N368S mutant. Tiwary and his co-authors discovered that the N368S mutation makes the protein so flexible that one of these pathways opens up, allowing Gleevec to leave the protein before it starts working.
Tiwary's method could also be reversed, using computer simulations to predict changes in a drug that would make it more effective and bind longer, which is a long-term goal of his lab. In another recent study published in ACS Central Science, Tiwary's simulations were used to predict which mutations might affect the interaction between RNA and small molecules. And while computer simulations may never replace traditional drug trials, this new study's designation as a "Hot Paper" shows that computational chemistry is earning its rightful place in the research world.
"For better or for worse, in chemistry there has been a mindset that simulations are perhaps not real chemistry. Sometimes this can be deeply entrenched," Tiwary said. "This work shows that simulations can complement experiments in ways that can eventually help us tackle problems of immense, worldwide societal relevance—for cancer and beyond."
More information: Mrinal Shekhar et al, Protein Flexibility and Dissociation Pathway Differentiation Can Explain Onset of Resistance Mutations in Kinases**, Angewandte Chemie International Edition (2022). DOI: 10.1002/anie.202200983
Yihang Wang et al, Interrogating RNA–Small Molecule Interactions with Structure Probing and Artificial Intelligence-Augmented Molecular Simulations, ACS Central Science (2022). DOI: 10.1021/acscentsci.2c00149
Provided by University of Maryland