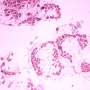
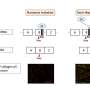
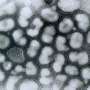
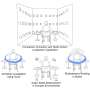
In vitro deletion of DMD exon 51 in a porcine DMDΔ52 kidney cell line by intein-mediated split-Cas9 system and subsequent nuclear transfer using the correctly modified cells, followed by embryo transfer to estrus-synchronized recipient sows. (B and C) Sanger sequencing (B) as well as agarose gel DNA electrophoresis (C) confirmed the absence of exons 51 and 52 in cDNA generated from mRNA of triceps brachii muscle samples in DMDΔ51-52 animals but not in DMDΔ52 and WT samples. (D and E) Immunohistochemical detection of dystrophin protein in the triceps muscle (D) and (E) myocardium. Bars = 100 µm. (F–I) Clinical chemical parameters in the blood serum: creatine kinase (CK; F), creatinine (G), alanine aminotransferase (ALT; H), and troponin I (I). The graphs show the median and the interquartile range (nWT = 5, n DMDΔ52 = 7, nDMDΔ51-52 = 5 for F–H; nWT = 4, n DMDΔ52 = 5, nDMDΔ51-52 = 5 for I). Significant differences are indicated: *P < 0.05; **P < 0.01; ***P < 0.001; and n.s. = not significant. Credit: Proceedings of the National Academy of Sciences (2023). DOI: 10.1073/pnas.2301250120")
Generation and characterization of DMDΔ51-52 animals. (A) In vitro deletion of DMD exon 51 in a porcine DMDΔ52 kidney cell line by intein-mediated split-Cas9 system and subsequent nuclear transfer using the correctly modified cells, followed by embryo transfer to estrus-synchronized recipient sows. (B and C) Sanger sequencing (B) as well as agarose gel DNA electrophoresis (C) confirmed the absence of exons 51 and 52 in cDNA generated from mRNA of triceps brachii muscle samples in DMDΔ51-52 animals but not in DMDΔ52 and WT samples. (D and E) Immunohistochemical detection of dystrophin protein in the triceps muscle (D) and (E) myocardium. Bars = 100 µm. (F–I) Clinical chemical parameters in the blood serum: creatine kinase (CK; F), creatinine (G), alanine aminotransferase (ALT; H), and troponin I (I). The graphs show the median and the interquartile range (nWT = 5, n DMDΔ52 = 7, nDMDΔ51-52 = 5 for F–H; nWT = 4, n DMDΔ52 = 5, nDMDΔ51-52 = 5 for I). Significant differences are indicated: *P < 0.05; **P < 0.01; ***P < 0.001; and n.s. = not significant. Credit: Proceedings of the National Academy of Sciences (2023). DOI: 10.1073/pnas.2301250120
A group of Ludwig Maximilian University scientists led by Eckhard Wolf has developed a porcine DMD model with a mutation that mimics the hallmarks of the human disease but develops them in an accelerated mode. The study is published in the journal Proceedings of the National Academy of Sciences.
Duchenne muscular dystrophy (DMD) is a fatal X-linked disease caused by mutations in the DMD gene, leading to complete absence of dystrophin and progressive degeneration of skeletal musculature and myocardium. The majority of DMD mutations are deletions of one or more exons, with a hotspot in the range of exons 45 to 55. Frame-shift mutations lead to the formation of a non-functional, truncated dystrophin or the complete absence of dystrophin due to premature stop-codons and—as a consequence—to severe muscular dystrophy. A prominent example is the loss of exon 52, which resembles one of the most frequent DMD mutations in humans.
The Wolf group has developed a porcine DMD model with this mutation, which mimics the biochemical, clinical and pathological hallmarks of the human disease but develops them in an accelerated mode. In DMD patients and in the corresponding pig model with a deletion of DMD exon 52 (DMDD52), expression of an internally shortened dystrophin can be achieved by skipping of DMD exon 51 to reframe the transcript. However, current delivery strategies for antisense oligonucleotides or gene editing tools limit exon skipping to a proportion of (cardio-)myocytes; the full therapeutic potential remains thus unclear.
Therefore, the Wolf group generated a model of ubiquitous correction of DMD by systemic deletion of exon 51 in the dystrophic pig model lacking DMD exon 52. Molecular, pathological, and clinical alterations were largely rescued, supporting the optimization of exon skipping strategies as a clinically relevant goal. Due to the high susceptibility of pigs to dystrophic muscle lesions, DMDD51-52 pigs resembling a form of Becker muscular dystrophy (BMD) will uncover long-term outcomes within a reasonable time frame.
This new study demonstrates that genetically tailored pig models are not only useful for studying disease mechanisms but also for predicting the best possible outcomes of targeted therapies.
More information: Michael Stirm et al, Systemic deletion of DMD exon 51 rescues clinically severe Duchenne muscular dystrophy in a pig model lacking DMD exon 52, Proceedings of the National Academy of Sciences (2023). DOI: 10.1073/pnas.2301250120
Journal information: Proceedings of the National Academy of Sciences
Provided by Ludwig Maximilian University of Munich