This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Researchers develop new tool for better classification of inherited disease-causing variants
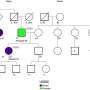
 Required input files for custom or CAVATICA workflow. (B) Variant classification method decision tree. (C) Alluvial plot showing how InterVar classification changes in AutoGVP. The large numbers of concordant VUS (357 656), LB (20 374), and B (28 310) variants were removed for easier visualization. Numbers in parentheses represent the number of variants in that classification. Panels (A) and (B) created with BioRender.com. Credit: Bioinformatics (2024). DOI: 10.1093/bioinformatics/btae114")
Researchers from Children's Hospital of Philadelphia (CHOP), the Perelman School of Medicine at the University of Pennsylvania, and the National Cancer Institute (NCI) of the National Institutes of Health have developed a new tool that allows scientists to annotate variant data from large-scale studies with clinically-focused classifications for risk of childhood cancer and other diseases. This new tool brings older applications in line with current guidelines and is available for use—for free—in the research community. The tool was described in a paper recently published in the journal Bioinformatics.
Whole genome and exome sequencing have become more widely available tools for clinical research in identifying inherited (germline) genetic variants that could result in a variety of diseases. While guidelines from the American College of Medical Genetics-Association for Molecular Pathology (ACMG-AMP) are frequently updated to help clinicians determine if germline variants are likely responsible for a patient's disease, automated tools may not always keep pace with such updates.
"Our goal was to create a publicly available tool that could evolve with these guidelines while still using many of the critical databases and approaches the research community has come to know," said senior study author Sharon J. Diskin, Ph.D., a member of the faculty of the Department of Biomedical and Health Informatics at CHOP and Associate Professor of Pediatrics at Penn Medicine.
The new tool, Automated Germline Variant Pathogenicity (AutoGVP), integrates germline variant pathogenicity annotations—information about whether identified variants cause disease—from the ClinVar database and sequence variant classifications from a modified version of the tool InterVar. AutoGVP returns pathogenicity classifications based on evolving ACMG-AMP guidelines through integration of ClinVar and InterVar. It addresses the InterVar tool's potential to overinterpret pathogenicity from loss-of-function variants that reduce the activity of a particular gene.
The need for AutoGVP became clear following a study published by Diskin and colleagues last year in the Journal of the National Cancer Institute that analyzed the germline DNA sequencing of 786 neuroblastoma patients and identified 116 pathogenic or likely pathogenic variants. The authors also reported that patients carrying these germline variants had a worse survival probability and identified BARD1 as an important neuroblastoma predisposition gene with both common and rare germline pathogenic or likely pathogenic variations.
A team including authors from the JNCI paper collaborated on the development of AutoGVP, to facilitate large-scale annotation of germline variants and assign pathogenicity automatically. They are applying AutoGVP to genetic data from pediatric brain tumor patients and larger neuroblastoma cohorts.
"Many samples we use for identifying pathogenic variants in pediatric brain tumors come from the centralized resource of the Children's Brain Tumor Network (CBTN), and we would like to be able to share new findings with the CBTN sites in a more streamlined manner," said Jo Lynne Rokita, Ph.D., a Supervisory Bioinformatics Scientist at the Center for Data-Driven Discovery (D3B) at CHOP and co-senior author of the study.
"With AutoGVP, we can streamline variant classification and swiftly incorporate new information as more and more biobanks release large sequencing data," said first author Jung Kim, Ph.D., a staff scientist at Division of Cancer Epidemiology and Genetics at the NCI. "Furthermore, AutoGVP reduces hands-on curating of variants and allows for reproducibility of the variant curation."
More information: Jung Kim et al, AutoGVP: a dockerized workflow integrating ClinVar and InterVar germline sequence variant classification, Bioinformatics (2024). DOI: 10.1093/bioinformatics/btae114