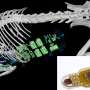
LoFreq detects single-nucleotide variants of E. coli bacteria with greater sensitivity than competing programs. Credit: Hemera/Thinkstock
A tumor is not a uniform mass of identical cells. However, teasing apart genetic heterogeneity within a biopsied tumor can be difficult. Researchers often fail to tell the difference between a rare variant in a DNA dataset or a small error because of imprecision in existing high-throughput sequencing technologies.
Now, a new computer program developed at A*STAR could help. Thanks to open-source software called LoFreq—so-called because it can detect mutations at extremely LOw FREQuencies—researchers can reliably pick out rare subpopulations of cells from heterogeneous populations of cancer cells, microorganisms and other biological samples.
"This is key to a wide range of scientific investigations, from understanding how pathogens evolve and escape the immune system, to uncovering the processes through which cancers grow and spread," says Niranjan Nagarajan, a senior scientist at the A*STAR Genome Institute of Singapore, who helped to develop the program.
Nagarajan and his co-workers wrote the algorithm that forms the foundations of LoFreq. Their aim was for the software not only to adapt to sequencing biases, but also to detect single DNA differences with frequencies below the specific level of noise introduced by sequencing errors. The researchers first tested the program against existing computer programs for analyzing large DNA datasets using simulated sequences from dengue virus. They then validated the approach using real genomic libraries from samples of Escherichia coli bacteria (see image), human gastric cancer biopsies, and dengue viruses collected before and after antiviral drug treatment—an exposure that often leads to the evolution of drug resistance in some subpopulations of virus.
"Previous attempts to describe this evolution have had to wait for the selection process to near completion," Nagarajan says. "In this new work, we have greatly increased the sensitivity of detecting these mutations and thus can catch their evolution in 'real time', observing how this process develops."
LoFreq proved itself to have near-perfect specificity for rare variants, with significantly improved sensitivity compared to existing methods, regardless of the high-throughput sequencing platform. The method also pinpointed a handful of low-frequency polymorphisms in whole-genome readouts from individual gastric cancer patients, and flagged mutational hotspots in dengue samples from a clinical drug trial.
"Almost anybody who is interested in studying evolutionary processes at a higher resolution, ranging from researchers who study how viruses and bacteria evolve and become more pathogenic, to cancer scientists looking at the evolution of a tumor," could benefit from LoFreq, Nagarajan says. The software is freely available via this link.
More information: Wilm, A., Aw, P. P. K., Bertrand, D., Yeo, G. H. T., Ong, S. H. et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Research 40, 11189–11201 (2012). nar.oxfordjournals.org/content/40/22/11189
Journal information: Nucleic Acids Research