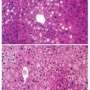
and is strongly stained red, indicating that it is full of fat. The sample on the right is from a mouse in which \"enzyme-dead\" HDAC3 is preventing the fatty liver. Note that there is much less red staining, even though the HDAC3 does not possess the enzyme activity for which it is named. Credit: Zheng Sun, Ph.D., Mitchell A. Lazar, M.D., Ph.D., Perelman School of Medicine, University of Pennsylvania")
A liver was stained for fat with Oil Red O. The sample on the left is from a mouse whose liver is depleted of histone deacetylase 3 (HDAC3) and is strongly stained red, indicating that it is full of fat. The sample on the right is from a mouse in which "enzyme-dead" HDAC3 is preventing the fatty liver. Note that there is much less red staining, even though the HDAC3 does not possess the enzyme activity for which it is named. Credit: Zheng Sun, Ph.D., Mitchell A. Lazar, M.D., Ph.D., Perelman School of Medicine, University of Pennsylvania
Drugs that inhibit the activity of enzymes called histone deacetylases (HDACs) are being widely developed for treating cancer and other diseases, with two already on the market. Researchers at the Perelman School of Medicine, University of Pennsylvania, show that a major HDAC still functions in mice even when its enzyme activity is abolished, suggesting that the beneficial effects of HDAC inhibitors may not actually be through inhibiting HDAC activity, and thus warranting the reassessment of the molecular targets of this class of drugs.
The study, appearing online in Molecular Cell this week, was conducted in the laboratory of Mitchell A. Lazar, M.D., Ph.D., director of the Institute for Diabetes, Obesity, and Metabolism. The Lazar lab has been working on HDAC3 for over a decade, focusing on the pivotal role of this enzyme in hormone-mediated regulation of gene expression and metabolism. They previously showed that depletion of HDAC3 in mouse liver upregulates expression of many genes involved in lipid synthesis, which causes a remarkable fatty liver. In the current study, they put "enzyme-dead" HDAC3 proteins back in the mouse liver and found, surprisingly, that the fatty liver can be rescued to a large degree.
Past studies showed that several other HDACs are intrinsically inactive and their enzyme activity is dependent on the activity of their cousin HDAC3. Many researchers thought the inactive HDAC enzymes worked by helping load HDAC3 onto its target molecule for catalyzing biochemical reactions.
The current study takes that scenario one step further, suggesting that enzyme activity may not be required for HDACs to execute their biological function after all. If this is true, the beneficial effects seen in drugs that inhibit HDAC activity may—despite what these drugs are called—not actually be through inhibition of HDACs.
The present study suggests that the name HDAC inhibitor can be misleading. "These drugs are so-named because they inhibit histone deacetylases, but we don't know what else they also inhibit," notes Lazar.
HDAC inhibitors bind zinc metal in the catalytic site of HDAC proteins. However, in addition to HDACs, the human body has nearly 300 enzymes that also depend on zinc and therefore are potentially also inhibited by HDAC inhibitors, notes leading author Zheng Sun, Ph.D., a postdoctoral fellow in the Lazar lab.
The study also raises questions about the role of histone acetylation in regulating gene expression. Histones are the proteins around which DNA is wrapped in the nucleus of cells. Acetate molecules get attached to histone, in the process of histone acetylation, which correlates with an increased expression of genes. HDACs, including HDAC3, remove these acetate groups, an action correlated with reduced expression of genes.
The Lazar group's study shows that this rule has exceptions. HDAC3 blocks the expression of the lipid-synthesizing genes and prevents fat build up in the liver. Investigators had thought that such repression happened by removing acetate groups from histones, which is suggested by the name HDAC3. However, by changing the DNA sequence of the natural form of HDAC3, the team now shows that enzyme-dead HDAC3 mutants are not able to perform histone deacetylation but are still able to repress gene expression and substitute for natural HDAC3 in preventing fatty liver.
These findings suggest that the acetylation reaction may not be the cause of active gene expression, or at least not sufficient to activate gene expression. Since HDAC inhibitors are currently in clinical use for cancer and are in trials for treatment of metabolic and neurological diseases, these new findings call for a re-thinking about how they work, say the authors.
Journal information: Molecular Cell
Provided by University of Pennsylvania School of Medicine