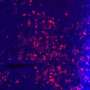
The molecular pathway for the protein p53, commonly associated with many cancers, is illuminated in this micrograph. The p53 - PIP2 complex is shown in red, DNA in blue, and the cell nucleus membrane in green. Unraveling the p53 - PIP2 pathway may lead to new therapies. Credit: Richard A. Anderson/UW-Madison
Scientists have long known that the protein p53, when mutated, is a critical factor in the onset of many different kinds of cancer. In its unmutated form, however, it is known to protect against cancer.
These dueling qualities make the p53 protein and the gene that makes it among the most studied in biology, yet the molecular mechanisms that govern its stability and function have yet to be fully understood.
Writing this week in the journal Nature Cell Biology, a team led by University of Wisconsin-Madison cancer researchers Richard A. Anderson and Vincent Cryns reports the discovery of an unexpected regulator of the critical protein, opening the door to the development of drugs that could target it.
"p53, like Janus, has two faces," says Anderson, referencing the Roman god of gates and doorways. "The p53 gene is the most frequently mutated gene in cancers, and when mutated it switches its function from a tumor suppressor to an oncogene that drives the majority of cancers."
Typically, explains Anderson of the UW School of Medicine and Public Health, the p53 protein serves as "the guardian of the genome," initiating the repair of DNA damaged by ultraviolet radiation, chemicals or other means and preventing tumor growth. When mutated, however, the protein goes rogue, becoming more stable and abundant than its unmutated counterpart, accumulating in the nucleus of the cell and causing cancer.
The research team, which includes study lead authors and postdoctoral fellows Suyong Choi and Mo Chen, found a new mechanism that drives this stability. The culprit: an enzyme called PIPK1-alpha and its lipid messenger, known as PIP2, which seem to behave as master regulators of p53.
The Wisconsin team showed that when a cell is stressed, whether by DNA damage or other means, the enzyme associates with p53 and produces PIP2, which binds strongly to it and promotes interaction between p53 and molecules known as small heat shock proteins. This stabilizes the protein complex, setting the stage for cancer, including aggressive cancers such as triple negative breast cancer.
"Small heat shock proteins are really good at stabilizing proteins," says Cryns, a professor of medicine in the UW School of Medicine and Public Health and expert on heat shock proteins. "In our case, their binding to mutant p53 likely facilitates its cancer-promoting actions, something we are actively exploring."
In the new study, the researchers also showed that when the PIP2 enzyme pathway is disrupted, mutant p53 does not accumulate and cause damage.
"If you can eliminate mutant p53, you might be able to eliminate cancers driven by p53," says Anderson.
The researchers are now actively searching for inhibitors of the PIPK1-alpha enzyme, a kinase, that could be used to treat tumors harboring p53 mutations.
"Although p53 is one of the most commonly mutated genes in cancer, we still do not have any drugs that specifically target p53," Cryns notes. "Our discovery of this new molecular complex points to several different ways to target p53 for destruction, including blocking the kinase or other molecules that bind to p53."
The findings, Anderson adds, were a little puzzling because the catalytic enzyme and PIP2 are typically and most abundantly found in cell membranes and not in the cell nucleus interior, where mutant p53 does its damage.
"This lipid pathway is weird," he says. "Lipids are supposed to be in membranes and this is not in a membrane compartment."
More information: A nuclear phosphoinositide kinase complex regulates p53, Nature Cell Biology (2019). DOI: 10.1038/s41556-019-0297-2, www.nature.com/articles/s41556-019-0297-2
Journal information: Nature Cell Biology
Provided by University of Wisconsin-Madison