Treatment that increases brain levels of an important regulatory enzyme may slow the loss of brain cells that characterizes Huntington's disease (HD) and other neurodegenerative disorders. In a report receiving advance online publication in Nature Medicine, a Massachusetts General Hospital (MGH)-based research team reports that increased expression of Sirt1, one of a family of enzymes called sirtuins, in the brain of a mouse model of HD protected against neurodegeneration. They also identified a potential mechanism for this protective effect.
"Diseases such as Huntington's, Parkinson's and Alzheimer's disease have different causative factors, but they share common themes – such as aggregation of misfolded proteins – and a unifying endpoint, the degenerative loss of neurons," says Dimitri Krainc, MD, PhD, of the MassGeneral Institute for Neurodegenerative Disease (MGH-MIND), the study's senior author. "Pharmacological targeting of Sirt1 may provide an opportunity for therapeutic development in HD and, more generally, in the neurodegenerative disorders of aging."
HD is an inherited disorder caused by a mutation in the gene for a protein called huntingtin. Deposits of the abnormal protein accumulate within the brain, causing destruction of brain cells. Symptoms of HD usually first appear in the middle years and worsen over the 10- to 30-year course of the disorder, leading to death from a variety of complications. Sirt1 is an important regulator of the activity of proteins involved in many critical functions – including energy metabolism, inflammation and stress tolerance – and recent studies have suggested it protects against the effects of several neurodegenerative diseases.
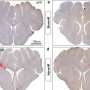
In experiments with a mouse model of HD, the researchers first showed that knocking out Sirt1 expression in the brain accelerated the appearance of HD-like pathology – such as aggregates of mutant huntingtin and increased cell damage in key areas of the brain. In contrast, a strain of HD mice in which Sirt1 was overexpressed lived longer, with significantly less neurodegeneration and huntingtin aggregation, than did HD mice in which Sirt1 expression was unaltered. Cellular experiments showed that Sirt1 overexpression directly protects neurons from the toxic effect of mutant huntingtin.
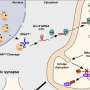
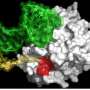
The MGH-MIND team also discovered a new target for Sirt1 activity in TORC1, a brain protein known to regulate several important neuronal genes, and found that the presence of mutant huntingtin interferes with the interaction between Sirt1 and TORC1, reducing expression of the regulated genes. In the same issue of Nature Medicine, a research team based at Johns Hopkins School of Medicine reports similar neuroprotective effects for Sirt1. Co-authored by members of Krainc's team, the Hopkins study demonstrated that mutant huntingtin inhibits the activity of Sirt1, leading to deregulation of multiple Sirt1 targets, in two additional HD mouse models.
"Development of therapeutic agents for neurodegenerative diseases requires an in-depth understanding of the mechanisms that link the underlying biology with the resulting neuronal dysfunction," says Krainc, an associate professor of Neurology at Harvard Medical School. "Developing and testing Sirt1 activators that protect against disorders like HD will require accurate information on Sirt1 activity in the normal and diseased brain. We hope our studies can contribute valuable data to that effort, which will require collaborations with NIH, with industry and with foundations such as the Cure Huntington's Disease Initiative, one of the supporters of this study."
Provided by Massachusetts General Hospital