New results from a study performed at the University of Helsinki suggest that genomic information from circulating influenza viruses can help in producing more efficient seasonal vaccines. The researchers were able to develop a simple approach for reliable real-time tracking and prediction of viral evolution based on whole-genome sequences of influenza viruses.
Influenza vaccines have been developed to prevent the yearly outbreaks of this respiratory illness and to protect people at risk from developing severe disease. In contrast to many other vaccines, influenza vaccines need to be reformulated each year because circulating influenza viruses continuously evolve. This evolution is due to the ability of viruses to accumulate new mutations in their genome. Especially critical are those genomic regions that encode viral proteins recognized by the human immune system.
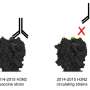
World Health Organization (WHO) is responsible for predicting the influenza strains that will be most common during the next season and for giving recommendations on the particular virus strains that should be used for producing the vaccines for the next influenza season. The effectiveness of the vaccines varies from year to year and was modest for the previous season. This could be due to antigenic mismatch between the circulating influenza viruses and the vaccine strains recommended by the WHO.
Researchers from the Institute for Molecular Medicine Finland, FIMM, the University of Helsinki, have now analysed thousands of complete genome sequences of influenza A(H1N1) and A(H3N2) strains representing different geographic regions. The study was done in collaboration with researchers from Singapore and United Kingdom. By utilizing biostatistical methods, the team was able to identify several genetic variants that changed the structure of the influenza proteins and that were present in surprisingly many of the strains studied.
"We named these characteristic variants as evolutionary markers as they seemed to contain valuable information regarding the viral evolution," explains Denis Kainov, the leader of the research team.
Furthermore, the group was able to show that both influenza subtypes acquired their own sets of these evolutionary markers. Importantly, many of these markers were not present in the virus strains used for the vaccine development during the last season.
"We believe that our results and methodology could further improve vaccine strain selection process and, thereby, enhance vaccine efficacy. Each year a substantial number of Finns gets influenza. We estimated that if the vaccination efficacy increases by 50%, this would save up to 8 000 people from getting the disease in Finland alone. Our team proposes using the strains containing all the identified evolutionary markers as vaccine candidates for upcoming influenza seasons," Kainov continues.
More information: Sergei S. Belanov et al. Genome-wide analysis of evolutionary markers of human influenza A(H1N1)pdm09 and A(H3N2) viruses may guide selection of vaccine strain candidates, Genome Biology and Evolution (2015). DOI: 10.1093/gbe/evv240
Provided by University of Helsinki