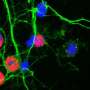
Credit: Fen-Biao Gao, PhD
A pair of collaborative studies led by Fen-Biao Gao, Ph.D., have identified two potential drug targets for the diseases amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). The studies, which appear in Nature Neuroscience and PNAS, provide a new layer of detail about how hexanucleotide repeat expansions in the C9ORF72 gene, the most common genetic mutation responsible for both ALS and FTD, causes neuron cell death. The Nature Neuroscience study also describes a new mouse model that more closely mimics the gradual build-up of toxins in patients with the diseases.
"Understanding how these mutations lead to motor neuron damage is important to the development of new treatment approaches," said Dr. Gao, the Governor Paul Cellucci Chair in Neuroscience Research and professor of neurology. "We know that this mutation can cause these diseases. These studies show that both mitochondrial function and DNA repair pathways are disrupted when the mutated gene is present in cells. That makes them potentially druggable targets."
In ALS, a progressive, neurodegenerative disorder affecting the motor neurons in the central nervous system, the C9ORF72 gene accounts for 40 percent of inherited forms of the disease and 6 percent of sporadic cases. As motor neurons die, the brain's ability to send signals to the body's muscles is compromised. This leads to loss of voluntary muscle movement, paralysis and eventually death from respiratory failure.
Similarly, FTD, originally called Pick's disease, is among the most common form of early-onset dementia, second only to Alzheimer's disease. FTD is caused by the loss of neurons in the frontal or temporal lobes.
Progress has been made in identifying genetic mutations that cause ALS and FTD, but how these changes affect neurons and cause toxicity is still poorly understood. Patients with the C9ORF72 mutation have an abnormally long GGGGCC repeat expansion in the gene. This expansion is responsible for the production of several unintended proteins called dipeptide repeat (DPR) proteins—ones not normally found in healthy people—that are in fact toxic to neurons, according to Gao and colleagues.
DNA repair pathway
In the PNAS study, Gao and his team found that the presence of DPR proteins disrupts the DNA repair machinery in neurons leading to the activation of programmed cell death. It turns out that fruit flies with the particular DPR protein known as poly(GR) produce too much of a protein called Ku80. A critical component of the DNA repair machinery, Ku80 is one of the first proteins to bind to a broken DNA strand. It then recruits the entire repair machinery needed to fix the break and reattach the two strands.
"We were surprised by these findings," said Gao. "Normally Ku80 is a good gene. Cells use it to fix DNA breaks. We were puzzled how a knock down of a good gene would rescue neurodegeneration in these flies."
Gao believes that as toxic levels of DPR proteins increase, this sends a signal that the cell is damaged and DNA repair is required. As a result, the cell produces more Ku80. As the mutated protein continues building up in the cell, more and more Ku80 is produced in response. Finally, the cell, thinking its damage is beyond repair, initiates programmed cell death and dies.
"It's like inflammation and bacteria," said Gao. "Inflammation is good because it tells the immune system to wake up and fight an infection, but too much inflammation can produce an adverse reaction. The cell is sending out this signal that it is damaged all the time and it keeps producing Ku80 that leads to overamplification of downstream signals such as P53. Eventually, the cell dies."
Reducing the amount of Ku80 reversed retinal neurodegeneration in fruit flies. It also reduced cell death signals in patient stem cell-derived motor neurons.
"This is potentially a great target for drugs," said Gao. "Relatively speaking, knockdowns are easier to target. Since we know it takes years for this toxicity to build it up, if we're able to start delivery early enough, it may be possible to temper this false signal in the cell, so it doesn't produce as much Ku80. This could potentially preserve motor neuron function."
Mitochondrial function
To better understand ALS/FTD disease mechanisms, Gao and colleagues developed a new mouse model in which DPR proteins gradually accumulate in mouse brain neurons. These DPR proteins are a product of the C9ORF72 mutation and the source of toxicity. In human neurons, they slowly build up over time. It takes years of exposure and accumulation before the neurons die. It's only later in life that ALS and FTD patients begin showing symptoms. This work is described in Nature Neuroscience.
"This is a much more realistic model for what we suspect is happening in human neurons with the C9ORF72 mutation," said Gao. "It takes years of build-up and stress for the toxicity to kill the cells."
Cells with the C9ORF72 mutation make many types of DPR proteins. Gao developed a mouse model specifically for the DPR protein, poly(GR). The poly(GR) gradually accumulates in the cortical excitatory neurons of the mouse. Using this model, trainees in the Gao lab found that the toxic polyGR protein binds to the ATP5A1 component of the mitochondrial complex.
Mitochondria are structures within cells that convert energy from food into a form that cells can use. Each cell contains hundreds to thousands of mitochondria that are located in the cytoplasm that surrounds the nucleus. The mitochondria take oxygen and simple sugars and create adenosine triphosphate (ATP), the cell's main energy source.
The ATP5A1 protein is critical to the production of ATP. When the mutant polyGR protein binds to the ATP5A1, it degrades. Gao believes the reduction in available energy output for the cell leads to early cellular defects and eventual neuronal cell death.
"It's possible that a drug, designed to boost mitochondrial function, might be able to offset some of the negative effects of the polyGR protein and rescue some neuronal function," said Gao. "Another strategy might be to boost ATP production, so the cell has enough energy to perform its functions."
The next step for Gao and colleagues will be to use their new mouse model to potentially identify drugs that hit either of these targets and assess whether that stops or reverses disease progression.
More information: So Yoen Choi et al. C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo, Nature Neuroscience (2019). DOI: 10.1038/s41593-019-0397-0
Rodrigo Lopez-Gonzalez et al. Partial inhibition of the overactivated Ku80-dependent DNA repair pathway rescues neurodegeneration in C9ORF72-ALS/FTD, Proceedings of the National Academy of Sciences (2019). DOI: 10.1073/pnas.1901313116
Journal information: Nature Neuroscience , Proceedings of the National Academy of Sciences
Provided by University of Massachusetts Medical School