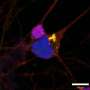
infiltrate the spinal cord as ALS disease progresses. Single molecule FISH detected IRF7 (an interferon-inducible gene) RNA molecules (red) in microglia and other cells. Credit: Valeria Gerbino/Maniatis lab/Columbia's Zuckerman Institute")
Representative confocal microscope image of the ventral horn of the spinal cord from an ALS mouse. Several microglial cells (green) infiltrate the spinal cord as ALS disease progresses. Single molecule FISH detected IRF7 (an interferon-inducible gene) RNA molecules (red) in microglia and other cells. Credit: Valeria Gerbino/Maniatis lab/Columbia's Zuckerman Institute
Columbia scientists have provided new insights into how mutations in a gene called TBK1 cause amyotrophic lateral sclerosis (ALS), a progressive neurodegenerative disease that robs patients of movement, speech and ultimately, their lives. The researchers found that ALS-associated mutations in TBK1 can have both positive and negative effects on the progression of disease in mice genetically modified to have ALS-like symptoms.
These findings, reported today in Neuron, provide both genetic and mechanistic insights that could lead to novel strategies for the development of treatments for ALS.
"One of the greatest challenges to finding a cure for ALS is using genetic data to understand the disease's underlying mechanisms," said the study's senior author, Tom Maniatis, Ph.D., a principal investigator at Columbia's Mortimer B. Zuckerman Mind Brain Behavior Institute. "For example, neurodegenerative diseases such as ALS involve multiple cell types that are constantly changing as the disease progresses, so a drug that is beneficial in the early stages of the disease could detrimental at later times. Our study provides a glimpse into the complex relationship between ALS genetics and its mechanisms, and highlights the challenge in developing safe and effective drugs to treat the disease," commented Dr. Maniatis, the Isidore S. Edelman Professor of Biochemistry at Columbia's Vagelos College of Physicians and Surgeons.
ALS is a disease of the spinal cord, which can be triggered by the accumulation of misfolded proteins in motor neurons, the nerve cells in the spinal cord that drive movement.
The human TBK1 gene is one of more than 25 genes that have been shown associate with or cause ALS when bearing a mutation. TBK1 is a central player in multiple cellular functions, including autophagy, a process that removes misfolded proteins, damaged bits of cells and bacterial pathogens. TBK1 is also required for the antimicrobial interferon response, which protects cells from bacterial and viral infections.
"TBK1 helps cells clear away clumps of misfolded proteins and defends the body against invading viruses and bacteria," said Valeria Gerbino, Ph.D., an associate research scientist in the Maniatis lab, and the paper's lead author. "And because individuals with certain mutations in TBK1 have ALS, we wanted to develop a deep, mechanistic understanding of how these mutations affect cellular functions in the spinal cord during the course of the disease."
Working in partnership with The Jackson Laboratory, Drs. Maniatis and Gerbino inserted ALS-causing TBK1 mutations in mice. These mice showed no signs of motor neuron disease.
"This was not surprising, as only a few of the many human ALS mutations tested thus far in mice by other ALS researchers have resulted in motor neuron disease," said Dr. Maniatis, who is also director of Columbia's Precision Medicine Initiative.
In an attempt to overcome this problem, the researchers inserted TBK1 mutations into an already established mouse model of ALS: the SOD1 mouse. Dr. Gerbino and collaborators at the Zuckerman Institute then tracked disease progression in these mice.
These new mice displayed striking differences in disease progression compared to mice that only bear the SOD1 mutations. Specifically, the disease onset began earlier.
"We found that mice bearing both SOD1 and TBK1 mutations experienced earlier movement abnormalities compared to those bearing only SOD1 mutations," said Dr. Gerbino. "A closer examination of the mice also revealed damage to motor neurons and the loss of muscle connections earlier than expected."
To further investigate this phenomenon, the scientists examined another group of SOD1 ALS mice, in which the TBK1 gene was deleted only in motor neurons, and not in other cells in the spinal cord. The team found that the absence of TBK1 activity in motor neurons alone led to the loss of autophagy in these cells. This loss led to an increase in the accumulation of harmful proteins, earlier death of motor neurons, and lost neuron-muscle connections. These observations demonstrated that early disease onset was a consequence of losing TBK1 in motor neurons.
Paradoxically, however, disease progression slowed down later in disease, and mice with TBK1 loss of function in all cells lived 25 percent longer than both the mice bearing only SOD1, as well as those mice for which the TBK1 gene was deleted only in motor neurons.
The team next investigated precisely how TBK1 mutations influenced other cells in the spinal cord. The researchers discovered that the interferon response was suppressed in glial cells, namely astrocytes and microglia. The interferon response, while critical for defense against infections in healthy people, can be toxic if over produced (as in the case of ALS mice), and can trigger a cascade of cellular processes that are toxic to motor neurons.
"The loss of TBK1 in microglia and astrocytes clearly diminishes the interferon response in the spinal cord of ALS mice," said Dr. Maniatis, who is Evnin Family Scientific Director and Chief Executive Officer at the New York Genome Center. "This correlates with significantly extending their lives."
"Our study provides an example of how multiple pathways within and between cells can be targeted through manipulation of a single gene, and how modifying these pathways in ALS can be beneficial—or detrimental—at different stages of disease progression," Dr. Gerbino added.
Dr. Maniatis argues that this type of foundational, discovery-driven research will move the needle toward treatments that focus on the underlying mechanisms driving ALS and its devastating symptoms.
"All neurodegenerative diseases ultimately begin with genes," said Dr. Maniatis. "As we continue to gather critical genetic information, we can identify mutations, like those in TBK1, which will provide additional mechanistic insights. These genes and the pathways they control will hold the key to developing drugs that make a difference in the life of ALS patients."
This paper is titled "The loss of TBK1 kinase activity in motor neurons or in all cell types differentially impacts ALS disease progression in SOD1 mice." Additional contributors include Esther Kaunga, Junqiang Ye, Daniele Canzio, Sean O'Keeffe, Noam Rudnick, Paolo Guarnieri and Cathleen Lutz.
Journal information: Neuron
Provided by Columbia University