Injection of virus-delivered gene silencer blocks ALS degeneration, saves motor function

Writing in Nature Medicine, an international team headed by researchers at University of California San Diego School of Medicine describe a new way to effectively deliver a gene-silencing vector to adult amyotrophic lateral sclerosis (ALS) mice, resulting in long-term suppression of the degenerative motor neuron disorder if treatment vector is delivered prior to disease onset, and blockage of disease progression in adult animals if treatment is initiated when symptoms have already appeared.
The findings are published in the December 23, 2019 online issue of the journal Nature Medicine. Martin Marsala, MD, professor in the Department of Anesthesiology at UC San Diego School of Medicine and a member of the Sanford Consortium for Regenerative Medicine, is senior author of the study.
ALS is a neurodegenerative disease that affects nerve cells in the brain and spinal cord. Motor neurons responsible for communicating movement are specifically harmed, with subsequent, progressive loss of muscle control affecting the ability to speak, eat, move and breathe. More than 5,000 Americans are diagnosed with ALS each year, with an estimated 30,000 persons currently living with the disease. While there are symptomatic treatments for ALS, there is currently no cure. The majority of patients succumb to the disease two to five years after diagnosis.
There are two types of ALS, sporadic and familial. Sporadic is the most common form, accounting for 90 to 95 percent of all cases. It may affect anyone. Familial ALS accounts for 5 to 10 percent of all cases in the United States, and is inherited. Previous studies show that at least 200 mutations of a gene called SOD1 are linked to ALS.
The SOD1 gene normally serves to provide instructions for making an enzyme called superoxide dismutase, which is widely used to break down superoxide radicals—toxic oxygen molecules produced as a byproduct of normal cell processes. Previous research has suggested that SOD1 gene mutations may result in ineffective removal of superoxide radicals or create other toxicities that cause motor neuron cell death, resulting in ALS.
The new approach involves injecting shRNA—an artificial RNA molecule capable of silencing or turning off a targeted gene—that is delivered to cells via a harmless adeno-associated virus. In the new research, single injections of the shRNA-carrying virus were placed at two sites in the spinal cord of adult mice expressing an ALS-causing mutation of the SOD1 gene, either just before disease onset or when the animals had begun showing symptoms.
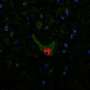
Earlier efforts elsewhere had involved introducing the silencing vector intravenously or into cerebrospinal fluid in early symptomatic mice, but disease progression, while delayed, continued and the mice soon died. In the new study, the single subpial injection (delivered below the pia matter, the delicate innermost membrane enveloping the brain and spinal cord) markedly mitigated neurodegeneration in pre-symptomatic mice, which displayed normal neurological function with no detectable disease onset. The functional effect corresponded with near-complete protection of motor neurons and other cells, including the junctions between neurons and muscle fibers.
In adult mice already displaying ALS-like symptoms, the injection effectively blocked further disease progression and degeneration of motor neurons.
In both approaches, the affected mice lived without negative side effects for the length of the study.
"At present, this therapeutic approach provides the most potent therapy ever demonstrated in mouse models of mutated SOD1 gene-linked ALS," said senior author Martin Marsala, MD, professor in the Department of Anesthesiology at UC San Diego School of Medicine.
"In addition, effective spinal cord delivery of AAV9 vector in adult animals suggests that the use of this new delivery method will likely be effective in treatment of other hereditary forms of ALS or other spinal neurodegenerative disorders that require spinal parenchymal delivery of therapeutic gene(s) or mutated-gene silencing machinery, such as in C9orf72 gene mutation-linked ALS or in some forms of lysosomal storage disease."
The research team also tested the injection approach in adult pigs, whose spinal cord dimensions are similar to humans, for safety and efficacy. Using an injection device developed for use in adult humans, they found the procedure could be performed reliably and without surgical complications.
Marsala said next steps involve additional safety studies with a large animal model to determine the optimal, safe dosage of treatment vector. "While no detectable side effects related to treatment were seen in mice more than one year after treatment, the definition of safety in large animal species more similar to humans is a critical step in advancing this treatment approach toward clinical testing."
More information: Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS, Nature Medicine (2019). DOI: 10.1038/s41591-019-0674-1 , nature.com/articles/s41591-019-0674-1