Body's own defense against ALS actually drives disease progression at later stages
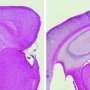
 in spinal interneurons.Right: autophagy-suppressed ALS models, showing virtually no aggregation. Credit: Tom Maniatis/Columbia University's Zuckerman Institute")
Columbia scientists have discovered that one of the body's natural defenses against amyotrophic lateral sclerosis (ALS)—a cellular 'clean-up process' called autophagy—suppresses disease progression early on, but in later stages advances the disease's deadly spread through the spinal cord. These findings in mice provide a window into ALS's earliest stages, as well as new insights into its complexity, namely the differing roles that autophagy plays in its progression. In addition, this study can help scientists search for ways to detect and even treat the disease before the onset of devastating symptoms that gradually rob patients of movement, speech and life.
The study is publishing this week in the Proceedings of the National Academy of Sciences.
"One of the biggest barriers to treating ALS is that its progression is dynamic—many different cell types and mechanisms are involved—so treating it at one stage of the disease might have very different, and potentially harmful, consequences at a different stage," said Tom Maniatis, PhD, a principal investigator at Columbia's Mortimer B. Zuckerman Mind Brain Behavior Institute and the study's senior author. "Here, we've identified a cellular process that likely plays a central role at the very beginnings of the disease, which could open the door to treatments that stop ALS before it has a chance to gain a foothold in the body."
ALS, also referred to as Lou Gehrig's disease, is a deadly disease of the central nervous system for which there is no effective treatment. It first attacks motor neurons, the type of nerve cells that guide muscle movement. As motor neurons die, disease spreads to other types of cells in the spinal cord. Over time, patients increasingly lose their ability to move, to speak and even to breathe on their own. In nearly all instances, patients succumb to ALS within five years of being diagnosed.
Despite the lack of effective therapies, however, much progress has been made in uncovering some of ALS's underlying mechanisms. For example, studies have shown that a common feature of all ALS patients is the abnormal accumulation, or aggregation, of protein clumps inside motor neurons. This accumulation kick starts autophagy, a critical cellular process in which such protein clumps, as well as other damaged parts of the cell, are broken down and removed. In addition, recent studies of ALS led by Columbia geneticist David Goldstein, PhD, revealed mutations in the genes that drives autophagy can cause ALS.
To more precisely explore the role of autophagy in ALS, Dr. Maniatis and his team developed two different groups of ALS mouse models. The first group was genetically modified to mimic the signs of ALS, but were otherwise normal. The second group was identical to the first but with one significant difference: autophagy in their motor neurons was suppressed. The researchers then compared how the disease progressed in these two groups. The differences they observed were unexpected. They found that, in the absence of autophagy, ALS symptoms in the second group of mice advanced more quickly early in the disease, but later on the spread of disease was slower. Remarkably, the mice with suppressed autophagy lived longer than the first group of ALS mice.
"This strongly suggested that even though autophagy may initially stave off disease by suppressing protein aggregation, it eventually hastens the spread of ALS and its devastating symptoms to the rest of the spinal cord," said Dr. Maniatis, who also directs Columbia's Precision Medicine Initiative and is the Isidore S. Edelman Professor and Chair of biochemistry & molecular biophysics at Columbia University Medical Center.
The authors argue that when autophagy is intact, other cell types that surround motor neurons undergo an inflammatory reaction, which soon spreads throughout the spinal cord, and contributes to the progression of ALS. However, in the absence of autophagy this spread occurs more slowly. They speculate that this may be due to the release of disease-causing protein aggregates that escape from dying motor neurons during the normal autophagy process. In the absence of autophagy this spread may be diminished.
Autophagy is governed by many genes working in concert across every cell in the body, so Dr. Maniatis and his team are now studying how mutations in these genes affect disease progression in their ALS mouse model.
"By the time ALS symptoms are noticeable, the disease is very far along so drugs aren't likely to work unless we can diagnose ALS much earlier—which will be helped by understanding how mutations in autophagy genes cause ALS and affect disease progression," said Dr. Maniatis. "If we can find ways of treating the earlier stages of disease, rather than trying to stop it after it's full blown, it may be possible to develop more effective therapies."
More information: Noam D. Rudnick el al., "Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS," PNAS (2017). www.pnas.org/cgi/doi/10.1073/pnas.1704294114