Study reveals gene critical to the early development of cilia
Researchers at the National Eye Institute (NEI) have described the functions of a gene responsible for anchoring cilia – sensory hair-like extensions present on almost every cell of the body. They show in a mouse model that without the gene Cc2d2a, cilia throughout the body failed to grow, and the mice died during the embryonic stage. The finding adds to an expanding body of knowledge about ciliopathies, a class of genetic disorders that result from defects in the structure or function of cilia. NEI is part of the National Institutes of Health.
The findings are published in the online journal Nature Communications. Senior author Anand Swaroop, Ph.D., is chief of the NEI laboratory of Neurobiology-Neurodegeneration and Repair. Lead author Shobi Veleri, Ph.D., is a research fellow in the laboratory.
Cilia are responsible for cell communication and play a key role in the receptor cells of sensory systems. For example, they are essential for odor detection in the nose and light reception in the eye. Because cilia are such a key element of cells, defects in genes that are involved in cilia development or function can cause complicated syndromes involving multiple organs and tissues
Bardet-Biedl and Joubert syndromes are examples of ciliopathies with symptoms that include deafness, kidney disease, and degeneration of the retina. Meckel syndrome is a ciliopathy so dangerous babies with the genetic defect rarely make it to term.
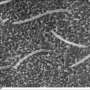
On individual cells, cilia grow from the basal body, a circular dent on the outer membrane acting as a platform. Supporting structures called distal and subdistal appendages, which are like the flying buttresses supporting Notre Dame Cathedral, anchor the platform in the basal body, priming it for the growth of cilia. Once anchored, the structures that form the cilium begin to extend from the site. Inside are a variety of proteins essential to maintain the cilium. Cc2d2a is believed to make a structural protein needed for cilia growth, but its precise functions have been unclear.
Researchers developed a mouse lacking Cc2d2a to investigate the gene.
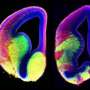
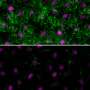
When they looked at the tissues of the mutant mice in very early stages of development, researchers found very few to no cilia, suggesting the gene plays a critical role at an early time. Looking closely at where the cilia should exist, the researchers saw that the supporting structures needed for cilia to grow were either completely missing or abnormal. In other experiments, the researchers found that the absence of Cc2d2a affected the activity of other genes and proteins involved in mouse nervous system development, including the key signaling protein, sonic hedgehog.
"This gene appears to play a key role in building structural support for the development of the cilia. Without this support, cilia are prevented from anchoring in the cell," said Dr. Swaroop. "It's like trying to build a house without a foundation. It's a big structural defect."
Studying the function and structure of cilia has become an active field of research that touches on many different organs and biological systems. More than 50 genes, including Cc2d2a have been discovered that, when defective, can lead to abnormal cilia development and ciliopathy in humans. By continuing to study how these genes work and interact, Dr. Swaroop said he hopes to gain further insight into not just how defects in genes related to cilia development in the retina cause vision problems, but the wider impact of these defects across body system and organs.
More information: Veleri, S et al. "Ciliopathy-associated gene Cc2d2a promotes assembly of subdistal appendages on the mother centriole during cilia biogenesis." Nature Communications, June 20, 2014. DOI: 10.1038/ncomms5207