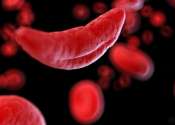
Sickle-cell disease (SCD), or sickle-cell anaemia (or anemia, SCA) or drepanocytosis, is an autosomal recessive genetic blood disorder with overdominance, characterized by red blood cells that assume an abnormal, rigid, sickle shape. Sickling decreases the cells' flexibility and results in a risk of various complications. The sickling occurs because of a mutation in the hemoglobin gene. Life expectancy is shortened. In 1994, in the US, the average life expectancy of persons with this condition was estimated to be 42 years in males and 48 years in females, but today, thanks to better management of the disease, patients can live into their 50s or beyond. In the UK, the current life expectancy is estimated to be 53–60 years of age.
Sickle-cell disease, usually presenting in childhood, occurs more commonly in people (or their descendants) from parts of tropical and sub-tropical regions where malaria is or was common. One-third of all indigenous inhabitants of Sub-Saharan Africa carry the gene, because in areas where malaria is common, there is a fitness benefit in carrying only a single sickle-cell gene (sickle cell trait). Those with only one of the two alleles of the sickle-cell disease, while not totally resistant, are more tolerant to the infection and thus show less severe symptoms when infected.
The prevalence of the disease in the United States is approximately 1 in 5,000, mostly affecting Americans of Sub-Saharan African descent, according to the National Institutes of Health. In the United States, about 1 out of 500 African-American children born will have sickle-cell anaemia.
Sickle-cell anaemia is the name of a specific form of sickle-cell disease in which there is homozygosity for the mutation that causes HbS. Sickle-cell anaemia is also referred to as "HbSS", "SS disease", "haemoglobin S" or permutations thereof. In heterozygous people, who have only one sickle gene and one normal adult haemoglobin gene, it is referred to as "HbAS" or "sickle cell trait". Other, rarer forms of sickle-cell disease include sickle-haemoglobin C disease (HbSC), sickle beta-plus-thalassaemia (HbS/β+) and sickle beta-zero-thalassaemia (HbS/β0). These other forms of sickle-cell disease are compound heterozygous states in which the person has only one copy of the mutation that causes HbS and one copy of another abnormal haemoglobin allele.
The term disease is applied, because the inherited abnormality causes a pathological condition that can lead to death and severe complications. Not all inherited variants of haemoglobin are detrimental, a concept known as genetic polymorphism.