Inventive approach may improve enzyme replacement therapy for Fabry disease
A new study uses a creative structure-based remodeling strategy to design a therapeutic protein that exhibits significant advantages over currently available treatments for a rare disease that often leads to cardiac and renal failure. The research, published by Cell Press on October 22nd in the American Journal of Human Genetics, describes a new and highly promising candidate for enzyme replacement therapy (ERT) for Fabry disease.
Fabry disease is a rare genetic disorder caused by a deficiency in alpha-galactosidase-A (GLA), an enzyme that breaks down fatty substances called glycolipids. Without the proper level of enzyme activity, a glycolipid called globotriaosylceramide (Gb3) accumulates to harmful levels inside cellular structures called lysosomes and damages the skin, nerves, eyes, kidneys and cardiovascular system. Although scientists have generated GLA for ERT, thus far this approach has proved challenging.
"Many patients have been successfully treated with these manufactured GLA proteins, but there are still problems to be resolved," explains senior study author Dr. Hitoshi Sakuraba from Meiji Pharmaceutical University in Tokyo. "For example, these enzymes are unstable in the blood, do not effectively reach the kidneys and heart and frequently cause an allergic reaction in Fabry patients."
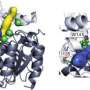
Dr. Sakuraba and colleagues took a different approach and, instead of making recombinant GLA, attempted to alter a different enzyme, called ?-N-acetylgalactosaminidase (NAGA), so that it could function like GLA. Normally, NAGA catalyses the hydrolysis of a different type of substrate and does not recognize the same substrates as GLA. Importantly, although NAGA is structurally similar to GLA, it does not react with the immune system in the same way.
The researchers examined the structures of GLA and NAGA and predicted how to alter NAGA so that it would recognize GLA substrates. Because the overall structure of NAGA was not changed, it was not expected to cause an allergic reaction in Fabry patients. The modified NAGA was found to be more stable than recombinant GLA and exhibited characteristics necessary for efficient incorporation into cells.
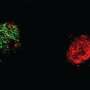
"Following confirmation of the effect of modified NAGA on cultured Fabry cells, we injected it into Fabry mice, and examined the incorporation of the enzyme into organs and its Gb3-degrading activity," explains Dr. Sakuraba. The modified NAGA was successfully incorporated into the liver, kidneys and the heart and there was a decrease in Gb3 accumulation in these organs.
"The enzyme has many advantages because of it high stability and the low possibility of the occurrence of an allergic reaction, although these characteristics should be confirmed in clinical studies in the future," concludes Dr. Sakuraba. "The modified NAGA is highly promising as a new enzyme for ERT for Fabry disease, and such structure-based designing of modified enzymes should be useful for the development of ERT for lysosomal storage diseases."
Source: Cell Press (news : web)