Researchers discover first gene linked to missing spleen in newborns
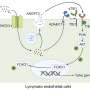
Researchers at Weill Cornell Medical College and Rockefeller University have identified the first gene to be linked to a rare condition in which babies are born without a spleen, putting those children at risk of dying from infections they cannot defend themselves against. The gene, Nkx2.5, was shown to regulate genesis of the spleen during early development in mice.
The study, published online May 3 in Developmental Cell, raises the hope that a simple genetic screening test for Nkx2.5 mutations can be developed that will alert parents that their developing child may be missing the organ, which could then be confirmed with a diagnostic scan.
"The great news is that with the appropriate preventive antibiotic treatment these children will not succumb to fatal infections. This test could potentially save lives," says the study's lead investigator, Dr. Licia Selleri, an associate professor in the Department of Cell and Developmental Biology at Weill Cornell Medical College.
Because defense against infections depends, in part, on the spleen, children known to be born without the organ require treatment with a regimen of antibiotic therapy throughout their lives. But most diagnoses of this condition, congenital asplenia, are made during an autopsy after a child dies, suddenly and unexpectedly, from a rapidly lethal infection, usually from bacteria that causes pneumonia or meningitis, Dr. Selleri says. "For those reasons, we believe this condition is not quite as rare as believed. Not every child who dies from an infection is given an autopsy."
Long search for genetic culprits
Patients with congenital asplenia usually lack a spleen as the sole abnormality, but sometimes have abnormalities of the heart and blood vessels. The majority of those cases arise sporadically, so are not believed to be inherited. One form of this disorder is known as Isolated Congenital Asplenia (ICA), characterized by a spleen that is missing but with no other developmental abnormalities. The cause is believed to be genetic, but no candidate genes in humans had been found before this study.
This research project was a collaboration between Dr. Selleri and her colleagues, and Rockefeller University's Dr. Jean-Laurent Casanova, professor in the St. Giles Laboratory of Human Genetics of Infectious Diseases. Dr. Casanova had led a previous study describing 20 ICA patients, of which most children suffered their first serious infection by age one, and nine died of an invasive pneumonia.
Dr. Selleri has long been studying congenital asplenia in the laboratory using the mouse as a model system and had previously discovered that a transcription factor known as Pbx is the prime regulator of spleen development in mouse models. Dr. Matthew Koss, a recent Ph.D. graduate who had studied in Dr. Selleri's lab, led the effort to create a strain of mice that lacked Pbx in the spleen, and were born without a spleen. He identified a regulatory module that is controlled by Pbx and targets Nkx2.5, a gene downstream of Pbx, in the developing spleen of the mouse embryo. He also discovered that Pbx controls the growth of the spleen by directly regulating the expression of Nkx2.5, which in turn controls cell proliferation within the primitive spleen organ.
Then, in Dr. Casanova's lab, Alexandre Bolze, a graduate student, sequenced genetic samples from ICA patients and analyzed them using whole exome sequencing technology, which allows sequencing of the entire coding genome of multiple patients -- a technique routinely employed by Dr. Casanova. Bolze found that Nkx2.5 was mutated in a family of asplenic patients, some of which died from lethal infections -- confirming the importance of Nkx2.5 in human congenital asplenia as in the mouse model of the disorder.
"This study illustrates the unique strength in using mouse models and human genetics hand-in-hand," says Dr. Selleri. "It demonstrates how genetic pathways identified in mouse models can be exploited to further understand the pathogenesis of human disease towards a better prenatal diagnosis."
She says that other patients and families with this disorder need to be studied in order to develop a comprehensive prenatal test. "It may be that there are other mutations that are acting in concert or independently of Nkx2.5 in other asplenic patients," Dr. Selleri says. Those studies in human patients are currently underway in the Rockefeller University lab, while at the Weill Cornell lab additional studies on mouse models are ongoing.