November 18, 2015 report
Hemolytic uremic syndrome: Search for the best treatment regimen with one of the world's most expansive medications
. Credit: Paulo Henrique Orlandi Mourao/ Wikipedia/ CC BY-SA 3.0")
(Medical Xpress)—Hemolytic uremic syndrome (HUS) is a severe illness that may lead to a complete loss of renal function. Most of the HUS cases are caused by infection with the shiga-toxin-producing Escherichia coli, well known from the 2011 epidemic in Germany, which resulted in 845 documented HUS cases and 54 deaths (1).
A rare, but more severe atypical form of HUS (aHUS) is not caused by the infection, but occurs spontaneously with over 50 percent of cases progressing to end stage renal disease (2). This form of the disease is caused by the attack of the body's own complement system on the capillary lining (endothelium) of glomeruli in the kidney.
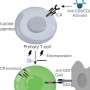
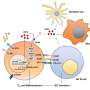
The complement system, a part of the innate immune system, consists of proteins circulating in the blood or bound to the cells. When complement is not active, its components are present in the form of inactive precursors. When complement is activated, the precursors are modified via proteolytic reactions. The primary goal of complement activation is elimination of pathogens. Complement can be activated via three pathways: the classical, the lectin and the alternative (Figure 1). While activation of the classical and the lectin pathways requires triggers, such as the presence of antibody-antigen complexes or carbohydrates from bacterial surfaces, alternative pathway can be activated spontaneously. The body's healthy cells are protected from complement attack by complement inhibitors.
Currently, alternative complement pathway deficiencies are identified in approximately 60% of aHUS patients (2, 3). These are either mutations in genes encoding alternative complement pathway proteins, or autoantibodies that impair the function of important complement regulator factor H (CFH).
Up until recently, treatment of aHUS patients has been an extremely challenging task (3). Plasma therapy has been used in which the patients receive plasma from healthy individuals to introduce normally functioning complement components. In some patients, this approach helps to preserve renal function, but it is insufficient in others. The nature of the disease also presents serious complications in case of aHUS-mediated renal failure. Most of the complement proteins, affected in aHUS, are produced in the liver. Thus, in the case of a renal failure and transplantation, there is a high risk (up to 100 percent) of aHUS recurrence in the graft. One way to circumvent this issue is to perform a double kidney-liver transplantation, which is highly risky for the patient.

In 2011, complement inhibitor eculizumab (trade name Soliris) was approved for the treatment of patients with aHUS, marking a new era in the treatment of this disease.
Eculizumab is a humanized monoclonal antibody directed against complement component C5 (Figure 1). During complement activation, C5 is split into C5a and C5b. Anaphylatoxin fragment C5a enhances permeability of blood vessels and mediates leukocyte migration via chemotaxis. On the other hand, C5b together with complement proteins C6, C7, C8 and C9 forms terminal complement complex (TCC), which normally causes lysis of pathogenic bacteria, such as Neisseria meningitidis, and in the absence of efficient complement control, can also lyse healthy cells. Eculizumab binds to C5 and prevents its activation into C5a and C5b, thereby blocking complement activation at this stage.
Although eculizumab is a very promising treatment for aHUS, it is very expensive. The cost, about €400,000 ($427,000) per year for an adult patient, puts enormous pressure on the health care system. The present Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines recommend life-long therapy with eculizumab infusions every two weeks (three weeks for the smallest infants) to prevent aHUS relapses. This recommendation has been questioned in clinical practice, mainly due to the following concerns: an increased risk of meningococcal infection and sepsis, possible development of neutralizing antibodies that may cause resistance to eculizumab, and the extremely high cost of this treatment.
Attempts have been made to discontinue therapy in recovered patients (4). In some cases, this led to a new aHUS episode; in others, it did not. It has been suggested that patients with certain mutations in the complement system (such as mutations in CFH) are at more risk of aHUS recurrence after discontinuation, but more studies should be performed to validate this hypothesis.
While the safety of complete therapy discontinuation remains an open question, optimization of treatment based on the individual patient's needs is clearly important. The current dosage regimen is adjusted to body weight only in pediatric patients weighting < 40 kg. All other patients receive the same dose; thus, a patient that weights 50 kg and a patient that weights 150 kg would be treated according to the same scheme.
Absence of the individualized approach is, partially due to the lack of fast and reliable tests to monitor the complement-inhibiting capacity of eculizumab in patients. Such tests should provide reliable data to determine whether complement is still fully blocked under the chosen treatment regimen (lower dosage or longer infusion intervals).
Recently, several in vitro methods have been published for the assessment of eculizumab blockade(5, 6). All of these methods are based on the fact that the complement activation can be triggered in serum samples in vitro by using specific antibodies, microbial components or cultured cells. Complement activation leads to measurable production of complement activation products, such as TCC. In the case of the complete eculizumab blockade, an increase of TCC levels upon activation is not observed. A recent study proposes an elegant method to estimate whether or not eculizumab is given to a patient in access(7). In this method, the patient sample is diluted with control human serum before activation. The results demonstrate that under the current dosage regimen, eculizumab was in excess for up at least 4 weeks after last infusion. This and other studies have indicated that extension of treatment intervals from two to four weeks may be possible.
In the future, the individualized approach to treatment, availability of generic drugs and extension of indication to other complement-mediated disorders may reduce the costs of the treatment and make complement inhibition more easily available to the patients.
More information: 1. Frank, C., D. Werber, J. P. Cramer, M. Askar, M. Faber, M. an der Heiden, H. Bernard, A. Fruth, R. Prager, A. Spode, M. Wadl, A. Zoufaly, S. Jordan, M. J. Kemper, P. Follin, L. Muller, L. A. King, B. Rosner, U. Buchholz, K. Stark, and G. Krause. 2011. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N Engl J Med 365: 1771-1780.
2. Mele, C., G. Remuzzi, and M. Noris. 2014. Hemolytic uremic syndrome. Semin Immunopathol 36: 399-420.
3. Davin, J. C., and N. C. van de Kar. 2015. Advances and challenges in the management of complement-mediated thrombotic microangiopathies. Ther Adv Hematol 6: 171-185.
4. Ardissino, G., S. Testa, I. Possenti, F. Tel, F. Paglialonga, S. Salardi, S. Tedeschi, M. Belingheri, and M. Cugno. 2014. Discontinuation of eculizumab maintenance treatment for atypical hemolytic uremic syndrome: a report of 10 cases. Am J Kidney Dis 64: 633-637.
5. Cugno, M., R. Gualtierotti, I. Possenti, S. Testa, F. Tel, S. Griffini, E. Grovetti, S. Tedeschi, S. Salardi, D. Cresseri, P. Messa, and G. Ardissino. 2014. Complement functional tests for monitoring eculizumab treatment in patients with atypical hemolytic uremic syndrome. J Thromb Haemost 12: 1440-1448.
6. Noris, M., M. Galbusera, S. Gastoldi, P. Macor, F. Banterla, E. Bresin, C. Tripodo, S. Bettoni, R. Donadelli, E. Valoti, F. Tedesco, A. Amore, R. Coppo, P. Ruggenenti, E. Gotti, and G. Remuzzi. 2014. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood 124: 1715-1726.
7. Volokhina, E. B., N. C. van de Kar, G. Bergseth, T. J. van der Velden, D. Westra, J. F. Wetzels, L. P. van den Heuvel, and T. E. Mollnes. 2015. Sensitive, reliable and easy-performed laboratory monitoring of eculizumab therapy in atypical hemolytic uremic syndrome. Clin Immunol 160: 237-243.
© 2015 Medical Xpress