Neuron type-specific gene loss linked to Angelman syndrome seizures
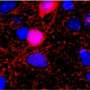
 of a human induced pluripotent stem cell-derived neuron. Credit: Thomas Deerinck, UC San Diego")
The gene UBE3A plays a critical role in early neurological development. If UBE3A is overexpressed - or if the enzymatic function of UBE3A protein is hyperactive - autism ensues. A lack of functional UBE3A causes Angelman syndrome (AS), a neurodevelopmental disorder characterized by severe developmental delay, motor deficits, absence of speech, and, in most cases, epilepsy. Prevention of seizures in AS model mice by restoring UBE3A after birth has proved largely unsuccessful, indicating that the timing of gene expression is also important.
And what about where UBE3A is expressed? That was the question UNC School of Medicine researchers wanted to answer.
In the journal Neuron, Ben Philpot, PhD, professor of cell biology and physiology, published his lab's research analyzing the spatial determinants for UBE3A loss in the development of seizures and other hyperexcitability phenotypes in the brain. Philpot's team used novel mouse models to show that selective UBE3A loss from GABAergic brain circuits - comprising neurons that release the inhibitory neurotransmitter GABA - led to the same kinds of seizure behaviors and electroencephalographic (EEG) abnormalities typically found in AS model mice, which lack UBE3A in all neurons.
Matt Judson, PhD, the study's first author and a research associate in Philpot's lab, said that the findings open the door for further investigations into the specific effects of UBE3A loss in other types of neurons.
"Our study has helped determine that UBE3A loss specifically from GABAergic neurons is what's critical for seizures in Angelman patients," Judson said. "But UBE3A loss from other neuron types may drive other phenotypes associated with the condition. This remains to be explored."
Given the inhibitory function of GABAergic neurons, the results are superficially unsurprising. GABAergic neurons transmit inhibitory signals that are necessary to balance the excitatory impulses mediated in large part by glutamatergic neurons. When this balance shifts too much in favor of excitation, epilepsy and other pathological outcomes can occur.
Previous studies indicated that glutamatergic neurons might play a bigger role in the development of seizures and epilepsy in AS. Philpot's lab showed that this is not the case.
Loss of UBE3A in glutamatergic neurons does impair their ability to receive certain inhibitory signals. Because these neurons are typically associated with excitation, this impairment would presumably promote hyperexcitability. Instead, the UNC researchers found that glutamatergic UBE3A loss alone was not sufficient to produce hyperexcitability.
"Our data indicate that impaired glutamatergic neurons don't drive the hyperexcitability," said Judson. "However, deficits in glutamatergic neurons could be related to other AS phenotypes."
Elucidating UBE3A functions within specific types of neurons may provide researchers with a clearer understanding of the molecular pathways both upstream and downstream, and may reveal new therapeutic strategies.
"Understanding the relevant cell types and regions for specific cell types should allow us to learn more about cellular pathways important for finding new drug targets," said Philpot, a member of the UNC Neuroscience Center.
While restoring UBE3A function in GABAergic neurons appears to be the most direct therapeutic strategy for the prevention of seizures in AS, it is still possible to reverse such symptoms in other ways.
"Previous research showed that it is much better to restore UBE3A function earlier than later, as the earlier you restore UBE3A the more phenotypes normalize," said Philpot. "However, we also know that there are other ways to overcome phenotypes that are not linked to UBE3A restoration. For example, seizures can still be treated in adults with anti-epileptics."