Mouse study links heart regeneration to telomere length
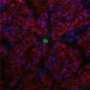
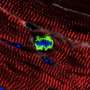
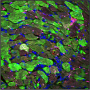
 has larger fibrotic regions (blue) compared with wild-type myocardium (left). Credit: Aix et al., 2016")
Researchers at the Spanish National Center for Cardiovascular Research have discovered that the ends of heart muscle cell chromosomes rapidly erode after birth, limiting the cells' ability to proliferate and replace damaged heart tissue. The study, "Postnatal telomere dysfunction induces cardiomyocyte cell-cycle arrest through p21 activation," which will be published online May 30, 2016 in The Journal of Cell Biology, suggests potential new interventions to boost the heart's capacity to repair itself after a heart attack.
Newborn babies can repair injured myocardium, but, in adults, heart attacks cause permanent damage, often leading to heart failure and death. Newborn mice can also regenerate damaged heart tissue. Their heart muscle cells, or cardiomyocytes, can proliferate and repair the heart in the first week after birth, but this regenerative capacity is lost as the mice grow older and the majority of their cardiomyocytes withdraw from the cell cycle.
Ignacio Flores and colleagues at the Spanish National Center for Cardiovascular Research (CNIC) in Madrid wondered whether the cause of this cell cycle arrest might involve telomeres, repetitive DNA sequences that protect the ends of chromosomes. If telomeres grow too short—due, for example, to a loss of the telomere-extending telomerase enzyme—cells can mistake chromosome ends for segments of damaged DNA, leading to the activation of a checkpoint that arrests the cell cycle.
Flores and colleagues therefore examined the length of telomeres in newborn mouse cardiomyocytes and found that the telomeres rapidly eroded in the first week after birth. This erosion coincided with a decrease in telomerase expression and was accompanied by the activation of the DNA damage response and a cell cycle inhibitor called p21.
Telomerase-deficient mice have shorter telomeres than wild-type animals, and, the researchers discovered, their cardiomyocytes already begin to stop proliferating one day after birth. When Flores and colleagues injured the hearts of one-day-old mice, telomerase-deficient cardiomyocytes failed to proliferate or regenerate the injured myocardium. In contrast, wild-type cardiomyocytes were able to proliferate and replace the damaged tissue.
They also found that knocking out the cell cycle inhibitor p21 extended the regenerative capacity of cardiomyocytes, allowing one-week-old p21-deficient mice to repair damaged cardiac tissue much more effectively than week-old wild-type animals.
Maintaining the length of cardiomyocyte telomeres might therefore boost the regenerative capacity of adult cells, improving the recovery of cardiac tissue following a heart attack. "We are now developing telomerase overexpression mouse models to see if we can extend the regenerative window," says Flores.
More information: Aix et al. 2016. J. Cell Biol. DOI: 10.1083/jcb.201510091