November 21, 2016 report
Finding your diagnosis in the brave new world of genetics-based medicine

(Medical Xpress)—We've done a number of articles recently about some amazing individuals developing personalized treatments to their own currently uncurable disease. Whether it is a rare orphan disease like Sanfilippo Syndrome and Castleman Disease, or something more common like cancer, these patients have been able to draw on their own medical expertise, or other economic and social resource to create well funded foundations and registries to marshal support for their cause.
There are countless other patients, however, who through age or profession don't have any pre-existing network to draw on for insider knowledge and proactivity. In many cases, particularly for the orphan diseases, these individuals don't even have a diagnosis they can use to advocate on their own behalf. Typically all they have is a stack of diffuse paper reports and xeroxed publications from the primary medical literature with no clear explanation to tie everything together.
To make matters worse, the often complicated genetics details they contain are not always immediately clear even to the experts. I know this now first hand, (and I am not a medical expert or professional) because I was given a set of of such reports by a neighbor and sought clear explanations from some of the best in business. This current state of affairs is not the direct fault of anybody in particular, but rather a side effect of an incomplete and evolving body of knowledge that necessarily contains significant ambiguity in its presentation.
Below I want to present the significant (at least as I have tried to understand them) genetic results for the case of Jackson Zuber, as given to me by his mother Emily. While obviously not intended to be a whole primer on the genetics, there should be enough detail so that we ourselves, and any professional geneticist, protein experimentalist or modeler, neurologist, neurobiologist, or radiologist clinician might extract the fuller picture and hopefully generate a few additional lines of inquiry.
Jackson is the person the geneticists designate as the 'proband', meaning the one who initiated the study, in this case a one year old boy. Exome sequencing revealed variants in four genes that were of significant clinical interest:
- NEB (nebulin) c.11450G>A; p.S3817N
- PLP1 (proteolipid protein 1) c.194T>G; p.I65S
- ERCC6 (excision repair cross-complementation group 1) c.2924G>A; p.R975Q
- PGAP1 (post-GPI attachment to proteins 1) c.2525+4C>T
The doctors logically focused on the PLP1 gene (and initially diagnosed the associated Pelizaeus-Merzbacher disease or 'PMD') because it is an X-linked homozygous gene. This means that Jackson only has the one copy of the gene, and would be particularly suseptibilty to any deleterious mutations in that gene . The other three genes are on 'autosomal chromosomes', heterozygous, and would therefore not immediately be prime suspects by virtue of the fact that another functioning copy of the gene is present.
That is not to say the other genes can be fully discounted, particularly given the absence of a full genome sequence which would contain any potential exome regions not analyzed in the exome sequence, including regulatory regions generally at the beginning of genes. It is also possible that one gene copy simply does not supply a required threshold level of the protein, or that the defective protein itself causing some new pathology.
It appears that the parental origin of all genes was determined (the so-called 'phase' analysis) but I do not know if that analysis determines whether the presumed 'good' copy of the gene could also have the same (and therefore undetected) variant.
Quite a bit is already known about the NEB gene and protein from many prior animal studies and although it is expressed in brains generally (in addition to the muscle regions where it is fairly critical), results from animals studies suggest there islargely normal cognitive function and neural structure in the presence of disabled nebulin genes.
PGAP1, technically heterozygous here, would only be an immediate red flag if there was also something like an undetected 'compound heterozygous' mutation (a second bad variant or polymorphism); in other words, the gene originating from the other presumed healthy parent has a different mutation, in which case serious neurologic issues are known to be a possible effect. PGAP1 is required for the production of GPI (glycosylphosphatidylinositol) that is attached to some proteins, and myelinating oligodendrocytes direct significant amounts these GPI proteins to the myelin sheath.

ERCC6 is involved in transcription coupled repair and similarly, it is typically associated with severe neurologic conditions only when both genes are affected (like in Cockayne syndrome). I only want to note here its implication in some microcephalic outcomes, including microcephalin and ATR (Seckle syndrome). If any significant question arises here one test for faulty ERCC6 repair capability might be a radiation sensitivity of skin fibroblasts, though I don't know how accurate and informative it would be.
In order to look closer at the specific case of Jackson's PLP1 variant we first need to decode and disambiguate the genetic notation for the variant; 'c.194T>G; p.I65S'. I will need to verify what I write here because errors are readily made.
The initial 'c.' indicates that we are looking at cDNA or complementary DNA, as we are dealing with exome sequencing info. It refers to an mRNA transcript's sequence expressed as DNA (GCAT) bases rather than as RNA (GCAU) bases. Having a 'genomic sequencing' reference (g.) would be a little more informative here for many reasons, namely, the presence of multiple transcription initiation sites (promoters), alternative splicing, the use of different poly-A addition signals, multiple translation initiation sites (ATG-codons), and the occurrence of length variations. Potentially, if exome sequencing draws on mRNAs after they are edited, (either in nucleus-specific or cytosol-specific editing), this would be an issue too, although RNA editing (post-transcriptional modification of bases, mostly A to G or A to I substitutions in humans) is quite rare.
As I understand the notation 194T>G, the 194th base pair position in Jackson's cDNA for PLP1 has a G while most normal cDNAs would have a T. Because G (like A) is a purine and T (like C) is a pyrimidine, this substitution is called a 'transversion' as opposed to a 'transition' (which would occur in the case of a purine to purine or pyrimidine to pyrimidine switch). Since there are natural mechanisms in the cell which more readily convert one-ring purines to other purines, or convert two-ring pyrimidines to other pyrimidines, transitions are significantly more common than transversions.
To get a better idea of how this T>G could have arisen I spoke with cell biologist Carl Smythe, a professor in the Department of Biomedical Science at University of Sheffield, and also geneticist Shane McKee, clinical director of the Belfast Health & Social Care Trust.The 'T>G' doesn't necessarily mean that a G has been directly changed into a T in the gene. For example, such a transversion could arise as a consequence of a mutation from a C to A on the non-coding strand. G-A bases can pair quite well (as do some others, although normal pairing is A to T and G to C) without causing major structural issues between the coding (sense) and noncoding (antisense) strands. As a consequence of this, the A would have a T inserted in opposite strand in the next round of synthesis.
Either strand could have had the original mutation, and the DNA replication process will give rise to two distinct coding sequences for a single locus. One can therefore end up with two cells each with complex phenotypes. After a non coding strand C to A mutation you get the stable G-A base pair, which after duplication gives a G-C pair and a T-A pair, where the latter corresponds to Jackson's mutation. This may have occurred during meiosis and have been in a sperm, or it may have occurred during development in whoever had the original mutation.
Because Jackson would have inherited his X chromosome with the variant PLP1 from his mom, the grandparents were checked and it was found that gramps had the same variant. Because gramps is asymptomatic the docs more or less recanted the PMD diagnosis. One possible explanation of this situation is that gramps could be a 'mosaic'. In other words the mutation was not present at the level of the sperm but rather arose later in development (as a somatic mutation), in which case it is possible that the cells that gave rise to gramps' nervous system have a normal copy of PLP1, and he is therefore quite normal. Another possibility is that gramps himself inherited the variant but none the less was able to repair it in the cells of his nervous system.
Although it would be rare, it is also conceivable that Jackson has the same mutation as gramps but it was independently gained, ie. it arose again in the bloodline as de novo variant in Jackson. Perhaps not totally inconceivable when you imagine that whatever genetic or metabolic background the gramps mutation originated in, a similar background would be expected to be present in Jackson. More typically the conventional thinking is that 'spontaneous' mutations arise more or less randomly during events like DNA synthesis when there is some non-negligible error rate during copying that escapes proofreading mechanisms.
It is also possible that the mutation does not have much effect in gramps' genetic background, but does have significant effect when occurring in the context of Jackson's genetic profile, ie. a 'facilitative' mutation necessary but not sufficient for PMD. One curious feature of PMD is that up to 70% of the patients have a duplicated PLP1 gene—an extra copy. It looks like this was explicitly checked for with Jackson, as exome sequencing wouldn't see it, but he did not have a duplication. His mitochondria were also sequenced and found to be normal, however in the face of not uncommon mitochondrial heteroplasmy (more than one unique set of mtDNA), we might also be curious what mitochondrial source was actually sampled here.
An important related question here is what tissue source got sequenced in the exome analysis—was it blood, skin, or epithelium? Because the same gene is typically spliced differently in different tissues it would also give different cDNAs in exome analysis of different tissues.
To get past this looming diagnostic roadblock, and in addition to whole sequence analysis, a functional protein study could be done to try and determine possible effects of the variant PLP1 substitution. This can include using software tools to model the structure and function of the protein, and actually constructing the variant protein in the lab and expressing it in animals to look for effects. For example, by creating what is called a conditional knock-in mouse line,

To start this kind of protein analysis one would need to look at the second part of the variant notation—the 'p.I65S'. Here the 'p' indicates we are talking about the amino acid sequence of the protein that corresponds to the cDNA or mRNA sequence. It says that Jackson's PLP1 will have a serine (S) substituted in at position 65 in place of the normal isoleucine.
If the possible isoleucine DNA codons (sense) are ATT, ATC, ATA, and the new variant possible serine codons are TCT, TCC, TCA, TCG, AGT, AGC, we can assume by process of elimination that it was the middle codon spot in either the ATT or ATC threonine that was changed into the AGT or AGC serine codon. I think it makes sense to presume that any base pair substitution generally originates and/or remains unrepaired in cells for some reason (even if that reason is excessive solar radiation applied to a skin cell), and that reason will typically reflect what is going on in the larger background metabolism and environment of the cell, and as it may happen, the organism.
Isoleucine is a hydrophobic amino acid and serine is a polar and uncharged amino acid. These are fairly different animals altogether and it is normally assumed that this kind substitution should have some significant effect on protein structure or function. The question is what effect? In checking some of the common software tools and databases for this kind of thing we find that 'PolyPhen2' says the substitution is probably damaging, 'MutationTaster' isn't happy with it either, and it is not recorded in either ExAC or 1000G.
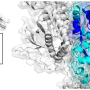
The canonical membrane structures of some of the various splice variants of the normal PLP1 protein have been determined well over a decade ago. It is a highly conserved protein that is virtually identical in several species from mouse to man. More recently, a few 3-D protein conformations, the actual crystal structures, have also been determined, sometimes in combination with other bound proteins. The presumptive membrane topology is four transmembrane helices, with the position 65 serine (or thereabouts depending on where the amino acid start count is done) lying at the extracellular apex of the first membrane helix. While serine can be phosphorylated in various proteins this may not be likely in the observed position.
As alternative splicing of the PLP gene yields four products—the classic PLP and DM20 proteolipids, and the more recently described proteolipids, srPLP and srDM20, it is important to try to understand how much of these various products are getting made by various kinds of cells in the nervous system, and their effects on those cells. A lot is already known, and more information is now acquired fairly continuously. Additionally, the (subcellular) localization of these products to various compartments within the cell is an important point (some get put into the myelin sheath, others get localized to the mitochondria, while other stay in the endoplasmic reticulum). The main question I think, at each instance, is whether there too much of this protein or not enough, and then also what is the effect of a poorly functioning, nonfunctioning or otherwise obstructive protein in each case? To this point, it is known that while transgenic mice that overexpress the PLP gene exhibit neuronal degeneration and axonal disintegration, perhaps paradoxically, the absence of PLP/DM20 in PLP null mice also causes axonal swellings. Because this protein is normally so abundant, around 50% of the total myelin protein, small changes can have large effects.
It is not known if the serine spot should affect splicing (but note nearby splice site in picture below), or affect any of the protein's cross-linked cysteines, or alternatively affect any critical cysteine palmitoylations, but further study would be needed. As the protein is also known to form dimers and maybe even higher older multimers, likely linking up to each other to across layers of compacted myelin, the effect of serine on such oligomerization may be an important question. Although most of those cysteines are closer to the beginning of the protein so to speak, they are a bit of an enigma these days because they can do so many things for the protein by virtue of their sulfur group. When cross linked or unlinked they change protein conformation, and also transduce ox-redox signals. When palmitoylated they target and localize the protein to the myelin sheath, and when spaced in various well defined 'localization sequences or motifs (like C-3xC, or C-10X, as here) they are also targeted to the mitochondria to participate in all kinds of functions.
It is critical I think to circulate Jackson's details to any doctors and researchers who might be poised to help, namely, experts in the various 'orphan' progressive degenerative neurologic disease. This class would include experts in various leukodystrophies and lysosomal storage diseases that affect myelin, and also those that ultimately affect mitochondria and their role in energy production and other key metabolic processes. Two guys who come immediately to mind, and who I have spoken to in the past for various articles are Bruno Benitez Washington University in St. Louis, and Doug Wallace at CHOP.
I will not delve much further into the other variants found in the genetic testing other than to note that the one for PGAP1 has a slightly different notation from the others, given as c.2525+4C>T. This annotation c.2525+4C>T appears to suggest that this variant is located +4 nucleotides apart from the last exonic nucleotide. This variant is predicted to be a "splice donor" which means that can alter the length of the resulting protein, a different transcript. PGAP1 has 22 exons and at least 11 splice variants. This variant has a mutation in the intron downstream of nucleotide position 2525. This creates a splice junction failure where the intron will not be spliced out and thus the variant will include protein sequence corresponding to the intron.
There is a lot of uncertainty about whether that would directly affect the enzyme activity of PGAP1. It will generate a protein of unexpected length. The precise number of splice variants affected by this mutation would need careful analysis, and the tissue sampled is likely to have a different spectrum of PGAP1 variants compared to brain. It is probably worth noting that PGAP mutations are associated with developmental defects, although again, much more information is clearly needed.
.
© 2016 Medical Xpress