Vet research identifies new target for taming Ebola
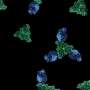
. Strong fluorescence at the periphery, or cell surface, and the filamentous projections are indicative of budding virus-like particles from the cell surface. Credit: PennVet Imaging Core (PVIC), directed by Bruce Freedman and managed by Gordon Ruthel")
Viruses and their hosts are in a eternal game of one-upmanship. If a host cell evolves a way to stop a virus from spreading, the virus will look for a new path. And so on and so forth.
A team of scientists led by Ronald Harty, a professor of pathobiology and microbiology at the University of Pennsylvania's School of Veterinary Medicine, has identified a mechanism that appears to represent one way that host cells have evolved to outsmart infection by Ebola and other viruses. In a new paper, he and colleagues reveal that host cells sequester viral proteins away from the plasma membrane within the cell, thus preventing viruses from spreading.
Harty says that finding a way to amplify this molecular interaction could lead to a novel antiviral strategy against Ebola that could temper the pathogen's damage.
"We think we have uncovered a cellular defense mechanism against Ebola and other viruses by which the cell can counteract the virus' ability to bud and spread to other cells," Harty said. "We want to dissect and understand this interaction further to potentially mimic the process for use as an antiviral strategy that could be used to treat these dangerous infections."
Jingjing Liang, the lead author and a doctoral student at Guangxi University who had a fellowship at Penn Vet to work in the Harty Lab for two years, performed the experiments in collaboration with Cari Sagum and Mark Beddford of the University of Texas at Austin, Sachdev Sidhu of the University of Toronto, Marius Sudol of the National University of Singapore and Ziying Han of Penn Vet.
The work appears this week in PLOS Pathogens.
Harty and colleagues have studied how viruses interact with host proteins with the long-term goal of finding ways to interrupt the virus's life cycle. In particular, they have examined the ways in which viruses hijack cell proteins to help regulate their exit and spread from the host cell through the budding process.
The scientists knew that, in infections with Ebola as well as many other viruses, including Marburg, rabies and HIV, viral matrix proteins, such as Ebola VP40, interacted with host proteins through short protein motifs: the PY motif on the viral protein and the WW motif on the host cell protein. Prior to this current study, all of the known interactions enabled the virus to bud efficiently from the cell.
In the current work, the researchers screened for new WW motifs from mammalian cell proteins that bound tightly to the PY motif of Ebola virus' VP40 protein. Not every PY motif binds to every WW domain; the interaction is specific, like a lock and key.
The screen turned up some proteins that the Penn team had explored before but also a new one, a protein called BAG3, known as a chaperone protein, which under normal physiological conditions acts to promote cell survival.
After confirming that Ebola VP40 interacted with full-length BAG3 in mammalian cells specifically via the WW domain, the researchers went on to test its functionality in influencing budding. They used a test that avoids manipulating the actual Ebola virus, which is too dangerous for the Penn Vet laboratory. Instead, they examined virus-like particles, which are produced by the virus's matrix protein, VP40, and are not infectious but accurately mimic the budding step of infection.
When the researchers examined cells expressing either Ebola or Marburg VP40 and then added in BAG3, they found that VLP budding went down in a dose-dependent manner. When they mutated the WW domain of BAG3 so it couldn't interact with VP40's PY domain, budding levels remained unchanged.
Knocking down levels of naturally occurring BAG3 with synthetic strands of RNA did just the opposite, increasing budding.
"With all of these assays, there was a consistent effect on budding levels, either up or down," Harty said.
Enhancing such an interaction, perhaps in combination with other therapies that attack the virus at other stages of its life cycle, could give the immune system the opening it needs to overcome an infection.
Using confocal microscopy and fluorescently labeled proteins, the scientists discovered that BAG3 appeared to be sequestering VP40 in the cell's cytoplasm away from the plasma membrane where the viral particles would need to go in order to bud off and spread to infect other cells. Their work identified BAG3 as the first WW containing host protein to negatively affect virus budding.
Although the group has not yet tested the VP40-BAG3 interaction with live Ebola or Marburg virus—those experiments are planned—they did find that BAG3 limits budding of a recombinant vesicular stomatitis virus that contains the Ebola PY motif.
"We used that to show that this works in a live virus infection," Harty said. "Taken together, we're hoping and assuming that it works the same way with the authentic Ebola virus."
In addition to testing BAG3 interactions with the Ebola and Marburg virus, Harty's lab also plans to further investigate what BAG3 is doing to VP40, whether it's simply sequestering it or whether it's modifying or degrading it.
And, while Harty noted that recent reports of a successful Ebola vaccine are undoubtedly good news, they don't lessen the need for a therapy.
"A vaccine is certainly important," he said, "but the therapeutics are a different arm of the antiviral strategy and are particularly essential for those who are already infected."
More information: Jingjing Liang et al, Chaperone-Mediated Autophagy Protein BAG3 Negatively Regulates Ebola and Marburg VP40-Mediated Egress, PLOS Pathogens (2017). DOI: 10.1371/journal.ppat.1006132