Studying a catalyst for blood cancers

Imagine this scenario on a highway: A driver starts to make a sudden lane change but realizes his mistake and quickly veers back, too late. Other motorists have already reacted and, in some cases, collide. Meanwhile, the original motorist - the one who caused the problem - drives on.
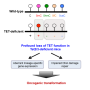
This is similar to what happens with the protein TET2 and a variety of blood cancers. TET2 is a tumor suppressor, preventing hematopoietic (blood) stem cells from overgrowing. However, if TET2 becomes mutated, which happens more frequently than we like, it allows other genes to mutate. TET2 loss does not actually create a cancerous state, but it helps create the conditions for cancer to thrive.
"If you lose TET2, it's not a malignant state, per se," said Mingjiang Xu, M.D., Ph.D., cancer researcher at Sylvester Comprehensive Cancer Center and associate professor of biochemistry and molecular biology at the University of Miami Miller School of Medicine. "But it's creating a situation for other mutations to happen, leading to all types of blood cancer."
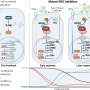
Xu and colleagues have been studying Tet2 for several years, and are starting to get a handle on how it operates. They published a paper today in the prestigious journal Nature Communications, which describes how TET2 loss can open the door for mutations that drive myeloid, lymphoid, and other cancers.
A different kind of mutation
That TET2 has a hand in several blood cancers makes it unique. Many mutated genes generate a specific type of cancer, depending on where they originate.
"If you lose TET2, it leads to blood cancers and it could be any type," said Xu. "Usually if you lose one gene, it leads to one specific cancer."
TET2 is an enzyme that demethylates DNA. Methylation turns down genes, keeping them from coding for specific proteins. In other words, TET2 may operate as a master switch, controlling whether certain genes are turned on or off.
TET2 mutations are found in 30 percent of myelodysplastic syndrome (MDS); 30 percent of secondary acute myeloid leukemias; and more than 50 percent of chronic myelomonocytic leukemias.
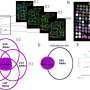
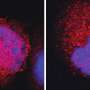
In the Nature Communications paper, Xu's team showed that mice without the Tet2 gene are more prone to blood cancers. In fact, removing Tet2 turns blood stem cells into mutation machines, and some of those malfunctions generate cancer.
Targeting Tet2
From a clinical standpoint, TET2 is a little tricky. First, it is easier to turn a protein off than turn it on. In addition, TET2 does not actually drive the cancer alone - it's the mutations acquired cooperate with the TET2 loss doing that nasty work. Turning up Tet2 could be helpful, but it has to happen early. Once the mutations are generated, targeting Tet2 would have little effect.
Still, Xu believes TET2 therapeutics could have a place in blood cancer treatment. He notes that around 5 percent of people over the age of 70 have TET2 mutations, which would make them ideal candidates for a preventive therapy.
"We are developing a method to target TET2," said Xu. "If we target that population for early therapy, we could potentially prevent those downstream mutations from happening."