Team characterizes the underlying cause of a form of macular degeneration

Named for Friedrich Best, who characterized the disease in 1905, Best disease, also known as vitelliform macular dystrophy, affects children and young adults and can cause severe declines in central vision as patients age. The disease is one in a group of conditions known as bestrophinopathies, all linked to mutations in the BEST1 gene. This gene is expressed in the retinal pigment epithelium, or RPE, a layer of cells that undergirds and nourishes photoreceptor cells, the rods and cones responsible for vision.
Despite the century of work on bestrophinopathies and the identification of genetic mutations responsible for the conditions, no one had identified the underlying mechanism that led to the vision loss seen in Best disease until now.
Using an animal model of Best disease in combination with biochemical and optical assays, a team of researchers at the University of Pennsylvania has pinpointed a number of abnormalities that give rise to the impairments seen in the disease.
"The genetic cause of the disease has been known for 20 years, but no one had samples of patients at the stage when the disease starts," said Karina E. Guziewicz, research assistant professor of ophthalmology in Penn's School of Veterinary Medicine and lead author on the study. But "we were now able to pinpoint this early stage and find out what factors trigger the development of lesions."
The new information sets the team up for testing a gene therapy to treat the disease, as the researchers will be able to observe whether or not these structural and biochemical abnormalities have been corrected.
"Now that we understand what we're seeing, it allows us to judge the success of a particular therapy," said Gustavo D. Aguirre, professor of medical genetics and ophthalmology at Penn Vet.
Kathleen Boesze-Battaglia, a professor in the Department of Biochemistry in Penn's School of Dental Medicine, also contributed her expertise in lipid biochemistry and spectral analysis of lipid debris to the study, which was published in the journal Progress in Retinal and Eye Research, the top ranked journal in the eye-research field.
"Interestingly, the lipid debris accumulation is similar to cholesterol rich plaque formation, compounded by a complexity of vitamin A metabolism," said Boesze-Battaglia. "Alterations in lipid metabolism likely contribute to the secondary disease pathology in this model."
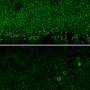
The main puzzle surrounding Best disease was why, despite the BEST1 gene being mutated in the RPE throughout the retina, vision loss struck the macula and fovea, the central areas of the retina responsible for reading and tasks requiring high-resolution vision, while seeming to spare the rest. Researchers had observed lesions in this area, but it was unknown why they developed.
In this study, the Penn-led team discovered that this predilection of the macula to develop lesions has to do with differences in the supporting structures of rods versus cones.
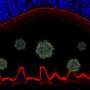
Rods, which make up more than 90 percent of photoreceptor cells in the retina and are responsible for dim-light vision, have a cluster of supporting structures known as RPE microvilli that cup the cell like stakes holding up a plant. In contrast, cones, the color-sensing photoreceptors that make up 3 to 5 percent of all photoreceptors but are overrepresented in the macula, are engulfed in a sheath of microvilli. In addition, cones are supported by an insoluble matrix.
Examining cross-sections of the fovea-like region in the canine macula of dogs affected with the canine equivalent of Best disease, the researchers found that the microvilli don't form and that the matrix is fragmented. The susceptibility of the macula is due to the fact that cones are the predominant cell type there and rely on the matrix for support and nutrient exchange.
"We were not expecting to find such dramatic structural abnormalities," Guziewicz said. "For a hundred years, this has been thought to be a disease of the RPE, but we have now identified this as a disease of the RPE-photoreceptor interface."
"The RPE provides transport of nutrients to the cones and engulfs the discarded part of cones and rods," said Aguirre. "When you lose the matrix, you lose the connection between those cells and the RPE and that leads to disease."
To determine if the same would be true in humans, the researchers looked at human induced pluripotent stem cell-derived RPE from Best disease patients and found similar signatures: microvilli numbers were decreased in length and density. These experiments were conducted in collaboration with David Gamm's laboratory from the McPherson Eye Research Institute at the University of Wisconsin-Madison.
Looking ahead, the research team would like to continue to probe the biochemical signals that lead to the improper development of the microvilli and matrix and push ahead with developing and testing a gene-therapy approach to treating bestrophinopathies.
"Knowing where the disruptions occur will allow us to develop proper outcome measures for a gene therapy, which is in the works," said Guziewicz.
More information: Karina E. Guziewicz et al, Bestrophinopathy: An RPE-photoreceptor interface disease, Progress in Retinal and Eye Research (2017). DOI: 10.1016/j.preteyeres.2017.01.005