New study offers hope for patients suffering from a rare form of blindness

A new form of therapy may halt or even reverse a form of progressive vision loss that, until now, has inevitably led to blindness. This hyper-targeted approach offers hope to individuals living with spinocerebellar ataxia type 7 (SCA7) and validates a new form of therapy with the potential to treat neurogenetic diseases effectively and with far fewer side effects than other medications. Details of this therapy appear in the latest issue of Science Translational Medicine.
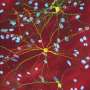
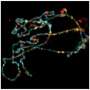
Senior author Al La Spada, MD, Ph.D., and colleagues treated mice with SCA7, an inherited neurodegenerative disease. Humans or mice with SCA7 have repetitions of the CAG triplet in the Ataxin-7 gene. These repetitions result in the production of Ataxin-7 protein with an expanded glutamine tract; the proteins then misfold and build up in retinal neurons, leading to their demise. La Spada's team evaluated a form of therapy known as antisense oligonucleotides (ASOs) for this condition. ASOs are synthetic, single-stranded pieces of DNA that bind to specific RNA molecules—in this case the RNA formed by the excess CAG repeat in the Ataxin-7 gene. The cell can then find and destroy the bound RNA molecule before it encodes the malformed protein that leads to visual loss.
ASOs offer a promising approach because they directly target the harmful proteins produced by genetic disorders before they can be expressed. Spinraza, a therapy now in use for patients with infantile-onset spinal muscular atrophy, uses a similar technique, and has been found to be effective in preventing motor neuron loss and improving symptoms in that condition.
The research team synthesized ASOs that matched with RNA produced by mouse Ataxin-7. Then, they injected the ASOs into the vitreous humor of one eye of SCA7 mice and a saline solution into the other eye. Eyes treated with the ASOs showed improved visual function compared to controls at both 4 and 6 weeks after treatment.
In assessing the therapeutic implications of this work, Dr. La Spada noted, "SCA7 is a rare disease, but is caused by accumulation of a misfolded protein, just like in Parkinson's disease or Alzheimer's disease. By developing ASOs to treat the retinal degeneration in SCA7, we are not only creating a powerful new therapy to treat this rare disease, but also will have an opportunity to evaluate this therapeutic strategy in a system (the eye) that is easily accessible and where the treatment response can be monitored with well-defined, objective read-outs."
The team also performed ophthalmologic evaluations on two human SCA7 patients and monitored the progression of retinal degeneration to better understand how the disease develops.
SCA7 is an autosomal dominant neurodegenerative disorder affecting about one in 500,000 individuals. It is one of nine recognized polyglutamine diseases caused by excess repetitions of the CAG nucleotide; others include Huntington's disease as well as six other forms of spinocerebellar ataxia. Individuals with SCA7 develop progressive neurologic and visual symptoms that appear anywhere from childhood to middle age. No treatments currently slow or stop the course of the disease.
More information: Chenchen Niu et al. Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7, Science Translational Medicine (2018). DOI: 10.1126/scitranslmed.aap8677