AI fast-tracks drug discovery to fight COVID-19

A global race is underway to discover a vaccine, drug, or combination of treatments that can disrupt the SARS-CoV-2 virus, which causes the COVID-19 disease, and prevent widespread deaths.
While researchers were able to rapidly identify a handful of known, Food and Drug Administration-approved drugs that may be promising, other major efforts are underway to screen every possible small molecule that might interact with the virus—and the proteins that control its behavior—to disrupt its activity.
The problem is, there are more than a billion such molecules. A researcher would conceivably want to test each one against the two dozen or so proteins in SARS-CoV-2 to see their effects. Such a project could use every wet lab in the world and still not be completed for centuries.
Computer modeling is a common approach used by academic researchers and pharmaceutical companies as a preliminary, filtering step in drug discovery. However, in this case, even every supercomputer on Earth could not test those billion molecules in a reasonable amount of time.
"Is it ever going to be possible to throw all of computing power available at the problem and get useful insights?" asks Arvind Ramanathan, a computational biologist in the Data Science and Learning Division at the U. S. Department of Energy's (DOE) Argonne National Laboratory and a senior scientist at the University of Chicago Consortium for Advanced Science and Engineering (CASE).
In addition to working faster, computational scientists are having to work smarter.
A large collaborative effort led by researchers at Argonne combines artificial intelligence with physics-based drug docking and molecular dynamics simulations to rapidly hone in on the most promising molecules to test in the lab.
Doing so turns the challenge into a data, or machine-learning-oriented, problem, Ramanathan says. "We're trying to build infrastructure to integrate AI and machine learning tools with physics-based tools. We bridge those two approaches to get a better bang for the buck."
The project is using several of the most powerful supercomputers on the planet—the Frontera and Longhorn supercomputers at the Texas Advanced Computing Center; Summit at Oak Ridge National Laboratory; Theta at the Argonne Leadership Computing Facility (ALCF); and Comet at the San Diego Supercomputing Center—to run millions of simulations, train the machine learning system to identify the factors that might make a given molecule a good candidate, and then do further explorations on the most promising results.
"TACC has been critical for our work, especially the Frontera machine," Ramanathan said. "We've been going at it for a while, using Frontera's CPUs to the maximum capacity to rapidly screen: taking virtual molecules and putting them next to a protein to see if it binds, and then infer from it whether other molecules will also do the same."
Doing so is no small task. In the first week, the team tested six million molecules. They are currently simulating 300,000 ligands per hour on Frontera.
"Having the ability to do a large amount of calculations is very good because it gives us hits that we can identify for further analysis."
Homing In On A Target
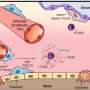
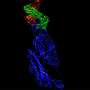
The team began by exploring one of the smaller of the 24 proteins that COVID-19 produces, ADRP (adenosine diphosphate ribose 1" phosphatase). Scientists do not entirely understand what function the protein performs, but it is implicated in viral replication.
Their deep-learning plus physics-based method is allowing them to reduce 1 billion possible molecules to 250 million; 250 million to 6 million; and 6 million to a few thousand. Of those, they selected the 30 or so with the highest "score" in terms of their ability to bind strongly to the protein, and disrupt the structure and dynamics of the protein—the ultimate goal.
They recently shared their results with experimental collaborators at the University of Chicago and the Frederick National Laboratory for Cancer Research to test in the lab and will soon publish their data in an open access report so thousands of teams can analyze the results and gain insights. Results of the lab experiments will further inform the deep learning models, helping fine-tune predictions for future protein-drug interactions.
The team has since moved on to the COVID-19 main protease, which plays an essential role in translating the viral RNA, and will soon begin work on larger proteins which are more challenging to compute, but may prove important. For instance, the team is preparing to simulate Rommie Amaro's all-atom model of entire virus, which is currently being produced on Frontera.
The team's work uses DeepDriveMD—Deep-Learning-Driven Adaptive Molecular Simulations for Protein Folding—a cutting-edge toolkit jointly developed by Ramanathan's team at Argonne, along with Shantenu Jha's team at Rutgers University/ Brookhaven National Laboratory (BNL) originally as part of the Exascale Computing Project.
Ramanathan and his collaborators are not the only researchers applying machine and deep learning to the COVID-19 drug discovery problem. But according to Arvind, their approach is rare in the degree to which AI and simulation are tightly-integrated and iterative, and not just used post-simulation.
"We built the toolkit to do the deep learning online, enabling it to sample as we go along," Ramanathan said. "We first train it with some data, then allow it to infer on incoming simulation data very quickly. Then, based on the new snapshots it identifies, the approach automatically decides if the training needs to be revised."
The system first establishes the binding stability of potential molecules in a fairly simple way, then adds more and more complex elements, like water, or performs finer analyses of the energy profile of the system. "Information is added at different funneling points and based on the results, it might need to revise the docking or machine learning algorithms."
Its complex workflows are carefully orchestrated across multiple supercomputers using RADICAL-Cybertools, advanced workload execution and scheduling tools developed by computational experts at Rutgers/ BNL.
"The workflows have sophisticated requirements," said Shantenu Jha, chair of BNL's Center for Data-Driven Discovery and the lead of RADICAL. "Thanks to TACC's technical support we were able to achieve both the desired levels of throughput and scale on Frontera and Longhorn within a couple of days and start production runs."
Applying The Weapons Of Science
The team had some advantages in getting their research off the ground.
The U. S. Department of Energy operates some of the most advanced X-ray crystallography labs in the world, and collaborates with many others. They were able to quickly extract the 3-D structures of many of the COVID-19 proteins—the first step in doing computational modeling to explore how such proteins respond to drug-like molecules.
They also were actively working on a project with the National Cancer Institute to use the DeepDriveMD workflow to identify promising drugs to combat cancer. They quickly pivoted to COVID-19 with tools and methods that had already been tested and optimized.
Though AI is frequently considered a black box, Ramanathan says their methods do not just blindly generate a list of targets. DeepDriveMD deduces what common aspects of a protein make it a better candidate, and communicates those insights to researchers to help them understand what is actually happening in the virus with and without drug interactions.
"Our deep learning models can hone in on chemical groups that we think are critical for interactions," he said. "We don't know if it's true, but we find docking scores are higher and believe it captures important concepts. This is not just important for what happens with this virus. We're also trying to understand how viruses work generally."
Once a drug-like small molecule is found to be effective in the lab, further testing (computational and experimental) is required to go from a promising target to a cure.
"Developing vaccines takes such a long time because molecules need to be optimized for function. They must be studied to determine that they're not toxic and don't do other harm, and also that they can be produced at scale," Ramanathan said.
All of these further steps, the researchers believe, can be accelerated by the use of a hybrid AI- and physics-based modeling approach.
According to Rick Stevens, Argonne's associate laboratory director for Computing, Environment and Life Sciences, TACC has been extremely supportive of their efforts.
"The rapid response and engagement we have received from TACC has made a critical difference in our ability to identify new therapeutic options for COVID-19," Stevens said. "Access to TACC's computing resources and expertise have enabled us to scale up the research collaboration applying advanced computing to one of today's biggest challenges."
The project compliments epidemiological and genetic research efforts supported by TACC, which is enabling more than 30 teams to undertake research that would not otherwise be achievable in the timeframe this crisis requires.
"In times of global need like this, it's important not only that we bring all of our resources to bear, but that we do so in the most innovative ways possible," said TACC Executive Director Dan Stanzione. "We've pivoted many of our resources towards crucial research in the fight against COVID-19, but supporting the new AI methodologies in this project gives us the chance to use those resources even more effectively."