Simple new testing method aims to improve time-release drugs

When you take a time-release drug, you count on it doing what the package says: release the drug slowly into your bloodstream to provide benefits over the specified period of time. When the drug dissolves too slowly or too quickly, the results can range from inconvenient— a decongestant that lets your sinuses get stuffed up too soon— to tragic, as many who were prescribed OxyContin discovered.
OxyContin, which contains the opiate oxycodone, was supposed to offer 12-hour pain relief. Instead, in some patients it dissolved much more quickly, causing them to take it more frequently and ultimately become addicted.
But assessing how a drug dissolves in the body is surprisingly tricky. Drug dissolution has to be measured under laboratory conditions that come as close as possible to mimicking what happens in the body.
In a paper published in Scientific Reports, UC Riverside researchers describe a simple, inexpensive way to measure drug dissolution that should help pharmaceutical companies develop better and more consistent time-release drug products.
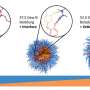
"We directly measured dissolution profiles of single drug granules, which are the little spheres you see when you open up a capsule," said corresponding author William Grover, an associate professor of bioengineering at the Marlan and Rosemary Bourns College of Engineering. "We accomplished this using a vibrating tube sensor, which is just a piece of glass tubing bent in the shape of a tuning fork."
Many factors affect drug dissolution in the body, including the pH and chemical composition of the gastrointestinal fluid, the hydrodynamics of the fluid caused by gastrointestinal contractions, the patient's sex, and metabolism. For example, the makers of OxyContin note taking the drug with a high-fat meal can increase the amount of oxycodone in the patient's blood by 25%.
Pharmaceutical companies usually test drugs by placing them in a vessel filled with fluid that mimics the contents of the gastrointestinal, or GI, tract, and stir the fluid to recreate GI tract dynamics. Small samples of the fluid are taken at intervals and the concentration of the drug, which should be increasing over time, measured using ultraviolet-visible spectroscopy or high-performance liquid chromatography. The data from this testing is used to construct a model of how the drug is expected to behave in the body.
The common ways of testing all have drawbacks. Small differences in the placement of tablets in a vessel can double the measured dissolution rate in one method, for example. Other methods can experience clogged equipment, impeded flow, and air bubbles, all of which affect how the drug dissolves. Moreover, the measurement process is time-consuming, laborious, often irreproducible, and involves expensive equipment. The existing methods also offer only "snapshots" of dissolution, taken at sampling points, providing limited information.
Grover, doctoral student Heran Bhakta, and undergraduate student Jessica Lin took a radically different approach. Rather than measure the increasing concentration of the drug in the fluid, they decided to measure the decreasing mass of a solid pellet as it dissolves.
The group used a glass tube bent like a tuning fork, kept vibrating by a circuit at its resonance frequency, which was determined by the mass of the tube and its contents. When they filled the tube with simulated stomach and intestine contents and passed an over-the-counter time-release drug granule through the tube, they observed a brief change in the frequency.
When plotted, they could compare the peaks of resonance frequency against the time to learn the buoyant mass of the drug granule at that moment.
"By passing the granule back-and-forth through the vibrating tube while it dissolves, we can monitor its weight throughout the dissolution process and obtain single-granule dissolution profiles," Grover said.
The group tested three different controlled-release proton pump inhibitor drugs: omeprazole, lansoprazole, and esomeprazole. Though they all have the same intended function in the body, they have very different granule sizes and dissolution mechanisms.
"We also found different dissolution behaviors between name-brand and generic formulations of the same drug. These differences in single-particle dissolution behavior could lead to different rates of drug absorption in patients," Grover said.
The researchers write that the technique addresses many of the shortcomings of existing testing methods, requires no additional analytical instruments, and is suitable for both fast-dissolving and slow-dissolving formulations. By giving dissolution profiles for individual pellets the method can capture variations in pellet dissolution behavior that other methods can't.
"Our technique is much cheaper and easier to perform than conventional methods, and that enables pharmaceutical companies to do more tests in a wider variety of conditions," said Grover. "We can also easily see differences in dissolution between individual particles in a drug. That should help pharmaceutical companies improve and monitor the consistency of their manufacturing processes."
The technique measures not just active ingredients, but also the inert ingredients in each drug particle.
"That's helpful for manufacturers who want to study how each layer of a controlled-release granule behaves during dissolution," said Bhakta.
The authors hope this data can augment existing dissolution methods and help pharmaceutical developers and manufacturers create better controlled-release drugs.
More information: Heran C. Bhakta et al, Measuring dissolution profiles of single controlled-release drug pellets, Scientific Reports (2020). DOI: 10.1038/s41598-020-76089-z