Researchers home in on a new cause of Stargardt disease
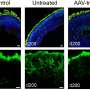
 Cell-surface-capturing (CSC) technology selectively purifies surface peptides on the apical and basal sides of primary human fetal RPE (hfRPE) by oxidation of N-glycan moieties followed by biotinylation proteolysis, affinity purification, and high-mass-accuracy mass spectrometry (n = 3). (B) Western blot analysis of ABCA4 in the membrane (M) and cytoplasmic (C) fractions and fibroblast (negative control). Na+/K+ ATPase, an apical membrane protein in RPE cells, is used as a positive control (n = 3 for each hfRPE, iRPE, and fibroblast). (C) iRPE monolayer cross-sections immune stained for ABCA4 (red) and Na+/K+ ATPase (green) (left panels) and for ABCA4 (red) and collagen IV (green) (right panels) (n = 3). (D) Transmission electron microscopy of immunogold-labeled (15 nm gold particles) ABCA4 in iRPE cells (n = 3). Arrowheads mark ABCA4 labeled with 15 nm gold particles in RPE apical processes. Inset shows higher magnification of apical processes containing gold particles. n represents independent samples from each group. Credit: Stem Cell Reports (2022). DOI: 10.1016/j.stemcr.2022.10.001")
Using a new stem-cell based model made from skin cells, scientists found the first direct evidence that Stargardt-related ABCA4 gene mutations affect a layer of cells in the eye called the retinal pigment epithelium (RPE). The discovery points to a new understanding of Stargardt disease progression and suggests a therapeutic strategy for the disease, which currently lacks treatment. The study took place at the National Eye Institute (NEI), part of the National Institutes of Health. The findings published online today in Stem Cell Reports.
"This new model will accelerate development of therapies for Stargardt disease," said NEI Director Michael F. Chiang, M.D. "We lack a therapy for this disease in part because it's rare. This model theoretically creates an unlimited supply of human cells for study." Stargardt affects about 1 in every 10,000 people in the U.S.
Stargardt disease causes progressive loss of central and night vision. The vision loss is associated with the toxic build-up of lipid-rich deposits in the RPE, whose main job is to support and nourish the retina's light sensing photoreceptors. Under normal conditions, the ABCA4 gene makes a protein that prevents this toxic build-up. Prior research showed that Stargardt disease is caused by a variety of mutations in the ABCA4 gene. More than 800 ABCA4 mutations are known to be associated with a broad spectrum of Stargardt disease phenotypes.
One way the RPE supports photoreceptors is by ingesting their spent outer segments, which keeps the cell pruned and healthy. In Stargardt disease, many scientists believe that RPE cells die after they acquire toxic byproducts when they ingest outer segments, and that this in turn leads to photoreceptor death and vision loss.
Much of the current understanding of Stargardt disease was gained by studying mouse models, which are inherently limited owing to the wide genetic variability of the disease in humans. With a human model of RPE, the NEI investigators were able to determine if ABCA4 gene mutations directly affected the RPE independent of photoreceptors.
To develop the model, the researchers took skin cells from Stargardt patients, converted them to stem cells, and then coaxed the stem cells to differentiate into RPE cells. Examining the patient-derived RPE, researchers detected ABCA4 protein on the RPE cell membrane. They explored the function of ABCA4 in RPE development by using the gene editing technology CRISPR/Cas9 to generate patient-derived RPE lacking ABCA4, called an ABCA4 knockout. They found that loss of ABCA4 did not affect maturation of the patient-derived RPE.
However, when the RPE lacking ABCA4 were exposed to normal (wild type) photoreceptor outer segments, the RPE cells accumulated intracellular lipids deposits.
Further tests of the ABCA4 knockouts showed evidence of defective RPE lipid metabolism and an impaired ability to digest photoreceptor outer segments, leading to lipid deposits in RPE cells.
This is the first report where loss of ABCA4 function in human RPE has been associated with intracellular lipid deposits in those cells, without exposure to ABCA4 mutant photoreceptor outer segments. Over time, these lipid deposits may contribute to RPE atrophy, leading to photoreceptor degeneration.
"Our report provides guidance for a gene therapy approach to target RPE," said the study's lead investigator, Kapil Bharti, Ph.D., senior investigator of the NEI Ocular and Stem Cell Translational Research Section. "Our data suggests that in addition to correcting ABCA4 loss of function in photoreceptors, gene therapies need to also target RPE cells."
This research is part of a larger effort by the NEI to address the limited availability of patient-derived stem cell lines for studying Stargardt disease. To overcome this barrier, the NEI initiated a STGD1-iPSC banking program from patients with different ABAC4 mutations. These cells will be made available to the community at-large for mechanistic and genotype-phenotype studies.
More information: Kapil Bharti, Cell Autonomous Lipid Handling Defects in Stargardt iPS Cell-Derived Retinal Pigment Epithelium Cells, Stem Cell Reports (2022). DOI: 10.1016/j.stemcr.2022.10.001. www.cell.com/stem-cell-reports … 2213-6711(22)00497-0