This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
trusted source
proofread
Researchers pursue three gene therapies for rare inherited disease

When neurobiologist David Corey showed up at a rare disease conference in 2017, he had no idea that he would enter a race against time to develop a treatment for it.
The conference was for Usher syndrome type 1F. Patients with this condition have a gene mutation that causes them to be born deaf and gradually lose their vision as they grow up. Corey, the Bertarelli Professor of Translational Medical Science in the Blavatnik Institute at Harvard Medical School, had devoted decades to studying the defective gene in a different context.
So when Corey happened upon an announcement for an Usher 1F conference in Boston, he knew he had to attend.
There, he introduced himself to Elliot Chaikof, chief of surgery at Beth Israel Deaconess Medical Center and the Johnson and Johnson professor of surgery at HMS, his wife Melissa, and their adult daughters, Rachel and Jessica, both of whom have Usher 1F. The Chaikofs had organized the conference through the nonprofit research collaborative they founded to find a treatment for the blindness part of the disease.
Meeting the Chaikofs—and especially Rachel and Jessica—stirred in Corey a powerful desire to help.
"We really felt that we know so much about this gene, if we don't try to do something for the disease, who else is going to?" Corey said.
Six years later, the Corey lab has three candidate gene therapies for Usher 1F blindness. Each takes a different approach to correcting the disease-causing mutation.
The researchers are now testing the therapies in animal models and are confident that at least one will move to clinical trials in humans to become a successful treatment.
Defining the problem
Usher 1F is a particularly severe form of Usher syndrome, in which a gene mutation causes cells in the eye and ear to stop producing an essential protein. Rachel and Jessica's gene mutation is most prevalent in the Ashkenazi Jewish community.
People with Usher 1F are usually born deaf and lacking the ability to balance. They develop an eye disease called retinitis pigmentosa, which causes a progressive loss of vision as the retina degenerates. Night vision often disappears first, followed by peripheral vision. For Jessica, this has meant giving up driving and getting a service dog to help her navigate the world around her. Eventually, people become completely blind.
"Everyone with Usher 1F has a unique experience, but one of the biggest challenges for me is slowly losing my independence as I lose my vision," Jessica said.
Like many people with Usher 1F, Rachel and Jessica have benefited from cochlear implants, which have improved their ability to hear and communicate. However, the Chaikofs learned soon after their daughters were diagnosed that there was almost no research on the condition and virtually no therapies to treat it—so in 2013, they established Usher 1F Collaborative.
"Essentially there were zero research groups working on this particular problem—nothing, nobody," Elliot said.
A call to action
While the Chaikofs were seeking answers, the Corey lab in the Department of Neurobiology at HMS was studying a protein called protocadherin-15 and its role in deafness.
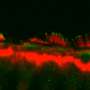
The researchers figured out that protocadherin-15 in the inner ear helps sensory receptors called hair cells convert mechanical vibrations into electrical signals, which the brain interprets as sound. Without protocadherin-15, the conversion doesn't happen, and the brain is unable to detect sound.
They also found protocadherin-15 in light-sensing cells, or photoreceptors, in the eye. However, they weren't sure of its exact function there, nor did they know why people lacking the protein in their eyes lose their sight over time.
In the course of the research, Corey had become aware that in Usher 1F, a mutation in the gene that makes protocadherin-15 causes cells in the ear and eye to stop producing it.
Connecting with the Chaikofs gave Corey a new motivation for his research and reinvigorated the family's quest for a cure.
"Because David understood the gene so well, he basically leapfrogged ahead of where the research was and hit the ground running," said Melissa, who is chair of Usher 1F Collaborative.
"For the first time we felt as if we had someone who could truly make a difference on this particular problem," Elliot added.
A daunting endeavor
The Corey lab chose to focus on the blindness aspect of Usher 1F, in part because patients are born profoundly deaf and lacking hair cells, making it unlikely that a therapy could restore their hearing. However, they are born with normal vision that gradually deteriorates, allowing time to intervene to preserve their sight.
The team found the perfect point person in Maryna Ivanchenko, an instructor in neurobiology at HMS who joined the lab to learn about gene therapy for deafness, but by serendipity is also an ophthalmologist and eye surgeon.
"Maryna has been absolutely central to this project because she understands both the hearing and the blindness aspects of this disease," Corey said.
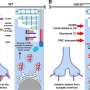
The researchers decided the way forward would be to design a gene therapy that either replaces or repairs the defective DNA that codes for protocadherin-15. Delivered inside eye cells, such a therapy would allow the cells to make the missing protein.
But the team encountered a major challenge: Protocadherin-15 is a huge protein, and the DNA that codes for it is too large to fit inside the usual delivery vehicle—a small capsule made from a non-infectious virus.
With the clock ticking for Rachel, Jessica, and thousands of others with Usher 1F, the team knew that their best hope for quickly developing an effective gene therapy would be to work on three different potential solutions at the same time.
Three shots on goal
The logistics of such a project are daunting.
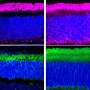
Because there is no good model for Usher 1F blindness in mice, Corey and colleagues started with a mouse model of deafness to test whether each therapy can restore protocadherin-15 production. They then moved to zebrafish, which offer a better model for progressive blindness. Testing continues in human eye cells grown in a dish, to be followed by tests in non-human primates. Only then can a therapy be deemed safe and effective enough for a human clinical trial.
"The project has been enormously complex because we're testing three different strategies in two different organs and four different species," Corey said. "Every time you add something, it multiplies the effort."
Even so, the Corey lab has made considerable progress on all three strategies:
- The most promising so far is the mini gene—a shortened but still functional version of the protocadherin-15 gene that easily fits inside the viral capsule that carries the DNA into a cell. In a recent study, the researchers showed their mini gene can restore hearing in mice, and early experiments in zebrafish suggest that it can also restore vision.
- The second strategy, called the dual approach, involves cutting the DNA in half. Each half is small enough to fit in the viral capsule. Once inside the cell, the halves reconnect, and can begin making the full-length protein. The approach has been used for other genes, Corey noted, and seems to be well suited for protocadherin-15. The researchers have tested this approach in mice and are starting to test it for safety in non-human primates.
- The final strategy involves putting gene editing tools into the viral capsule instead of loading it with a replacement gene. Once inside the cell, the gene editors would correct the protocadherin-15 gene mutation. Many mutations cause the condition, but the researchers are designing the tools to fix the most common one, R245x. The researchers recently demonstrated that their approach can counteract hearing loss in mice.
Corey and his collaborators, including Marcos Sotomayor at The Ohio State University and Artur Indzhykulian, HMS assistant professor of otolaryngology head and neck surgery at Massachusetts Eye and Ear, hope at least one of these therapies will safely stop and perhaps even reverse vision loss in patients with Usher 1F.
"This is an outstanding example of how basic science can be translated to therapies," Elliot said. "The progress that David has made in a very short period of time has been remarkable."
The lab has plans to partner with a biotech company that will continue working on any therapies that succeed in initial testing.
Finding hope
If any of the strategies clear the hurdle of animal testing, the next step would be a clinical trial. Yet therein lies another challenge: Because Usher 1F is an orphan disease—meaning it affects fewer than 200,000 people in the United States—it will be hard to enroll enough people in a randomized controlled trial.
To get around this challenge, Usher 1F Collaborative has initiated a natural history study on vision loss in people with Usher 1F. The study will follow participants over four years to assess how their vision loss progresses naturally, essentially establishing the control group for a clinical trial ahead of time. That way, if and when a trial does happen, all participants can receive the therapy.
"We're on a parallel track with David's research so that when the natural history study is completed, we'll be ready for the clinic," Melissa said.
It's not clear which, if any, of the therapies will succeed, nor when it will happen, but the possibility that they could make a tangible difference in people's lives keeps Corey, Ivanchenko, and others motivated as they work long hours in the lab.
And the Corey lab's three shots on goal have become many: Eight other groups at universities in the United States, Canada, and Australia are also working on treatments for Usher 1F.
"There are a lot of patients and families whose world is about to go dark, and they need everyone's best ideas," Elliot said.
"I keep telling myself that there will be a time when I can finally stop letting my life revolve around Usher," Jessica said. "Being able to see the research is what keeps me sane; it's what helps me get through the day."