Solving mystery of how sulfa drugs kill bacteria yields 21st century drug development target
More than 70 years after the first sulfa drugs helped to revolutionize medical care and save millions of lives, St. Jude Children's Research Hospital scientists have determined at an atomic level the mechanism these medications use to kill bacteria. The discovery provides the basis for a new generation of antibiotics that would likely be harder for bacteria to resist and cause fewer side effects.
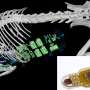
The work focused on sulfa drugs and their target enzyme, dihydropteroate synthase (DHPS). Most disease-causing microorganisms need DHPS to help make the molecule folate, which is required for the production of DNA and some amino acids. Working with enzymes from gram-negative and gram-positive bacteria, researchers used a variety of techniques to determine for the first time the key intermediate structure DHPS forms during the chemical reaction to advance folate production. The structure also explains at a molecular level how sulfa drugs function and how resistance causing mutations help bacteria withstand them.
The findings mark a major advance in both microbial biochemistry and anti-microbial drug discovery. The study is published in the March 2 issue of the journal Science.
"The structure we found was totally unexpected and really opens the door for us and others to design a new class of inhibitors targeting DHPS that will help us avoid side effects and other problems associated with sulfa drugs," said Stephen White, Ph.D., chair of the St. Jude Department of Structural Biology and the paper's corresponding author.
Co-author Richard Lee, Ph.D., a member of the St. Jude Department of Chemical Biology and Therapeutics, added: "Now we want to leverage this information to develop drugs against the opportunistic infections that threaten so many St. Jude patients."
Sulfa drugs were discovered in the 1930s and became the first antibiotic in widespread use. Although the drugs were early victims of antibiotic resistance, they are still widely used against emerging infectious diseases and to prevent infections in patients with weakened immune systems, including St. Jude patients undergoing cancer chemotherapy. The growing problem of antibiotic resistance has prompted renewed interest in sulfa drugs as a possible source of new therapeutic targets, Lee said.
Previous work had shown that sulfa drugs target DHPS and work by mimicking a molecule called pABA. DHPS advances folate production by accelerating the fusion of pABA and another molecule called dihydropteridine pyrophosphate (DHPP). Until now, however, scientists did not know exactly how the DHPS reaction occurred or how sulfa drugs disrupted the process.
Working on enzymes from gram-positive Bacillus anthracis and gram-negative Yersinia pestis, the bacteria that cause anthrax and plague, researchers first used computational methods to predict the enzyme's activity. Next they used a technique called X-ray crystallography to capture the unfolding chemical reaction and confirm the prediction. X-ray crystallography involves bombarding proteins trapped in crystals with X-rays to determine the protein structure.
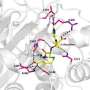
Researchers showed that DHPP binds to a specific pocket in DHPS. Aided by magnesium, the binding promotes the break-up of DHPP and release of pyrophosphate. Two long flexible loops then create an intermediate structure that sets the stage for pABA to enter and bind in a second short-lived pocket, allowing pABA to fuse with the cleaved DHPP. Investigators captured all four actors in the drama in a single crystal structure, including the intermediate cleaved DHPP molecule whose existence was previously unknown.
The results showed that the mechanism involves a chemical reaction known as an Sn1 reaction rather than the anticipated Sn2 reaction. "This is a key finding for drug discovery because it reveals chemical features of the DHPS enzyme's active site that we can exploit in developing new drugs," said study co-author Donald Bashford, Ph.D., an associate member of the St. Jude Department of Structural Biology.
The study also provided insights into sulfa drug resistance. Investigators showed that the binding sites of pABA and the sulfa drugs overlap, but that sulfa drugs extend beyond the pocket in which pABA binds. Mutations associated with drug resistance cluster around this extended region of the pABA pocket, which explains how mutations can prevent the drugs from binding without seriously affecting the binding of pABA. The work also highlights the transitory structure made by the two DHPS loops as a target for a new class of drugs that would be difficult for bacteria to develop resistance against.
"When we set out on this project eight years ago, a goal was to truly understand the catalytic mechanism of the DHPS protein and how the inhibitors targeting it work. I am ecstatic we've succeeded," Lee said. The success grew out of an interdisciplinary effort and some luck, White said. The plague enzyme turned out to be well suited to this project. Unlike the DHPS enzymes from other bacteria, the two extended loops are free to form the short-lived structure and the pABA pocket when the enzyme is immobilized in the crystal.
More information: "Catalysis and Sulfa Drug Resistance in Dihydropteroate Synthase," by M.-K. Yun et al. Science (2012).