Informatics approach helps doctors, patients make sense of genome data
The cost of sequencing the entire human genome, or exome – the regions of the genome that are translated into proteins that affect cell behavior – has decreased significantly, to the point where the cost of looking at the majority of a patient's genomic data may be less expensive than undertaking one or two targeted genetic tests. While efficient, the acquisition of this much genetic data – in some cases as many as 1.5 to 2 million variants – creates other challenges.
In a paper that appears today in the advance online edition of Genetics in Medicine, researchers from the University of North Carolina at Chapel Hill unveil an analysis framework aimed at helping clinicians spot "medically actionable findings" from genetic tests in an efficient manner.
"The challenge for medical geneticists is what do we do with the 'incidentalome' – the large amount of genetic data that these tests generate which may be important but which was incidental – that is, had nothing to do with why the patient underwent DNA analysis in the first place," said Jonathan Berg, MD, PhD, assistant professor of clinical genetics and a member of UNC Lineberger Comprehensive Cancer Center.
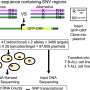
"Our team is faced with this issue in a clinical trial we are conducting called the NC GENES study. So we put together a framework that classifies genetic variations into three different 'bins': those that are linked to a treatable or preventable condition (the medically actionable); those that have a known link to conditions for which we don't have treatment options; and those for which there is no known direct association between a genetic variation and a disorder," he said.
The team then created an informatics approach to carry out a structured analysis on these three 'bins'.
"While there are still some challenges, we believe that this approach facilitates the analysis and streamlines the ability of the molecular analyst to go through a lot of data very quickly, providing more timely results to physicians and patients," says Berg.
Berg notes that the researchers had to set a very high bar for the genetic variants reported to patients and physicians, taking into account that there are errors in all of the current databases of known disease-causing mutations and that they contain variants that are probably not disease causing, due to unavoidable errors in data processing and other aspects of genetic research. However, because most hereditary disorders are very rare, disease causing mutations are highly unlikely.
"In epidemiologic terms we valued specificity over sensitivity. We will have some false negatives because we are ignoring some genetic variants that we don't understand well or that are very unlikely to occur. However, as researchers who also work with patients, we know that there are significant consequences to false positive results for genetic disorders and given the rarity of many of these disorders we think this is an appropriate risk," he argues.
Berg and his collaborators, which include James Evans, MD, PhD, Bryson Distinguished Professor of Genetics Research and a member of UNC Lineberger, are also studying the practical consequences of our ability to pinpoint disease-causing mutations in the genome.
"We hope that this methodology will enhance our ability to quickly translate a large amount of data into findings that are useful to physicians and patients, allowing us to study important issues like patient preference for learning about their likelihood of developing or passing along a hereditary disease for which there is no treatment," said Evans.
"These are important ethical considerations, and currently there exist no best practices because this technology is still relatively new," he added.