Using gene therapy to tackle complex brain disease
")
Substantial progress has been made in the development of treatments for a particular brain disease, thanks in part to an EU-funded project. The X-ALD project focused on achieving a better understanding of the pathogenic mechanisms leading to 'X-linked adrenoleukodystrophy', a disorder which results in the accumulation of long chain fatty acids in tissues throughout the body but especially in the central nervous system.
Most importantly, this research could open the door to the further development of novel therapeutic approaches. Researchers found for example that X-ALD could be treated with a combination of gene therapy and blood stem cell therapy.
'The main results of this project were the improved understanding of the molecular mechanism of X-ALD,' explains project coordinator Johannes Berger. 'Although we still don't fully understand the detailed mechanism, we have nonetheless moved substantially forward, something which is visible by the many papers published in journals such as Science and Nature Medicine. In addition, a novel strategy for the treatment of patients with an inflammatory form of X-ALD and have no transplant donor has been developed.'
X-ALD is one of the most common monogenetic disorders affecting the central nervous system (CNS). Ultimately the myelin sheath - the protective layer that coats the nerves - is destroyed, causing neurological problems. Patients who have no myelin succumb to mental and physical problems triggered by nerves that lose their function.
All forms of X-ALD are characterised by neurological abnormalities, which in the most severe cases can be fatal. While X-ALD is characterised biochemically by the accumulation of fatty acids, the role of this acid accumulation in the pathophysiology of the disease has remained a mystery. However, the X-ALD team has made substantial progress in the identification of candidates for modifier genes that could account for the phenotypic variability in the expansion and characterisation of X-ALD.
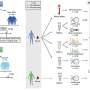
The gene affected in X-ALD codes for a protein that is probably involved in the transport of very long-chain fatty acids into a specific intracellular organelle, called the peroxisome. To this end, the project team developed a new system in which the properties of ALD protein can be studied under in vivo conditions using baker's yeast, as well as an in vitro system.
'X-ALD is a rare disease, and very little was known about the molecular mechanism underlying it,' explains Professor Berger. 'By bringing together experts in the field, we managed to achieve a major breakthrough in our understanding of the disease, and opened up novel therapeutic strategies that are now in clinical use.'
With respect to gene therapy, this project has moved even beyond the original scope to the first clinical application of gene therapy in X-ALD. A major impact of the work carried out in this project is expected with regard to future therapeutic treatment of X-ALD patients within the next 10 years.
Professor Berger believes that intense discussions and regular meetings enabled the project team to achieve focus. 'We were able to drive each other forward, and this was the main reason for the success of the project,' he says. 'The group has continued to work together, and has focused on other forms of the disease where there are still no treatments available.'
More information: X-ALD www.x-ald.nl