Research identifies cell receptor as target for anti-inflammatory immune response
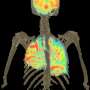
 with mice that are genetically engineered to lack certain receptors in their spleen (right), scientists have shown SIGNR1 receptors (blue) are required to facilitate an anti-inflammatory action.")
(PhysOrg.com) -- Invading pathogens provoke a series of molecular heroics that, when successful, muster an army of antibodies to neutralize the threat. Like with any close-quarter combat, however, an aggressive immune response runs the risk of friendly fire accidents. For the last decade, immunologists have intensively studied mechanisms evolved by the immune system to avoid these accidents by shutting off the immune response once the invaders have been eliminated.
Now the discovery of a new role for a specialized cell receptor has revealed aspects of how the immune system prevents a harmful overreaction to a foreign threat. Researchers at The Rockefeller University found that a receptor known to shield HIV and Hepatitis C from an effective immune response is also essential to the therapeutic effects of a common anti-inflammatory drug, intravenous immunoglobulin (IVIG). The finding opens up new possibilities for developing drugs to suppress the inflammation caused by autoimmune diseases such as rheumatoid arthritis and lupus.
“I see the implications as quite immediate,” says Jeffrey Ravetch, Theresa and Eugene M. Lang Professor and head of Rockefeller’s Laboratory of Molecular Genetics and Immunology. “We can develop new classes of anti-inflammatory molecules that can exploit this pathway. These findings also explain why certain pathogens like the HIV virus and Hepatitis C have usurped this pathway with their own mechanisms for evading host response.”
The research further demystifies the workings of IVIG, which have baffled scientists for years. Essentially, IVIG is a very high dose of the same class of antibodies — immunoglobulin cells called IgG — that perpetrate autoimmune diseases in the first place. Ravetch and colleagues in his lab partially solved this paradox in earlier work that identified a single sugar molecule called sialic acid at the tail end of some IgG molecules. When present, the sugar gives IgG molecules anti-inflammatory activity. If absent, the IgG molecules lose their protective qualities and become pro-inflammatory agents. Building on that finding, Ravetch in April published research in Science explaining how to engineer a molecule of sialylated IgG that — when given to arthritic mice — was 30 times more effective than standard IVIG treatment.
The latest findings, to be published as Ravetch’s inaugural paper in the Proceedings of the National Academy of Sciences, pushes this research further to define a special cell receptor that is required for IVIG to work. In mice, this receptor — SIGN-R1 — is found in a group of cells in the spleen that regulate the immune response in part by recognizing that special sugar found on some IgG molecules. “This recognition of and binding with the sialylated IgG cells seems to be the first step that is triggered by IVIG to suppress inflammation,” Ravetch says.
Ravetch and his colleagues homed in on the spleen by breeding transgenic mice lacking in key types of immune cells, dosing them with IVIG, and measuring whether it protected them from an arthritis-inducing agent that they were then exposed to. They found that mice were not protected when certain types of immune cells common to the spleen were deleted. A series of biochemical tests on the different receptors within those cells identified SIGN-R1 as the one that bound specifically to the molecules that help along the anti-inflammation response, sialylated IgG.
The findings should apply to humans, too. Ravetch and his colleagues identified a receptor in human cells — DC-SIGN — that behaves exactly as the SIGN-R1 found in mice. In our case, the receptors are found on dendritic cells, a prominent cell-type of the human immune system.
Now that he knows both the protein and receptor that initiate the immune response, Ravetch wants to develop molecules that can regulate that response. He also wants to know what, exactly, the sialylated IgG causes to happen that ultimately leads to the anti-inflammatory response. “It’s exciting to have this new pathway to dissect,” Ravetch says.
Provided by Rockefeller University