New therapeutic strategy could target toxic protein in most patients with Huntington's disease
Howard Hughes Medical Institute researchers have designed tiny RNA molecules that shut off the gene that causes Huntington's disease without damaging that gene's healthy counterpart, which maintains the health and vitality of neurons. Laboratory studies suggest that a single small interfering RNA could reduce production of the damaging Huntingtin protein in nearly half of people with the disease. Another 25 percent of patients might benefit from one of a set of four additional small interfering RNAs.
Phillip D. Zamore, an HHMI investigator at the University of Massachusetts Medical School in Worcester, and his colleagues reported their findings in an article published April 9, 2009, in the journal Current Biology.
There is no treatment for Huntington's disease, which is caused by a mutant form of the Huntingtin gene. Huntingtin is required for healthy nerve cells, but the mutant gene makes a toxic protein that contains excess amounts of the amino acid glutamine.
The key to whether the Huntingtin gene is normal or defective lies in a kind of genetic stutter: a repetitive sequence of the DNA triplet CAG, which codes for the amino acid glutamine. Stretches of CAG "repeats" appear in every human being's Huntingtin gene, but the length varies. Whereas the normal gene has a sequence of between six and 34 CAG repeats, the abnormal gene contains many more. In fact, any stretch of DNA containing more than 40 of these repeats ensures that its bearer will develop Huntington's—the greater the number of repeats, the earlier the disease strikes and the greater its ferocity. The abnormal Huntingtin protein causes movement disorders, cognitive failure, and ultimately, death. Children who have a parent with Huntington's disease have a 50 percent chance of inheriting the disease themselves.
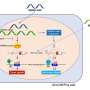
Zamore studies how RNA interference can be used to silence genes selectively. In the 1990s, he and other scientists learned they could shut down the production of specific proteins by introducing double-stranded RNA into the cell that is identical to the RNA they wanted to turn off. These strands of RNA, known as short interfering RNA (siRNA), slice apart the original RNA, which the cell then destroys.
But nine years ago, when researcher Neil Aronin, who is also at UMass Medical School, proposed using the technique to attack Huntington's, Zamore couldn't see a way.
"I explained to him that you can't," Zamore said. The problem was that the disease gene and its healthy allele are almost identical, and Zamore told Aronin that he wouldn't be able to distinguish between the two forms of Huntingtin. "Then, as he was leaving my office, it occurred to me that you could," he recalled. The key was something called a single nucleotide polymorphism or SNP.
A SNP is any place on the genetic code that varies by a single unit. The genetic code is written with four letters, A, C, T, and G, which stand for the four nucleotides, adenosine, cytidine, thymidine, and guanosine. The pattern of these nucleotides dictates which protein is encoded by a given gene. DNA in the nucleus is transcribed as messenger RNA, which leaves the nucleus and begins making proteins based on the order of these four bases. A person's two copies of any gene may vary at these locations "simply because the two parents have different ancestries," Zamore said.
Zamore, Aronin, and their collaborators decided to look for such variation in the Huntingtin gene. It was a bit of a long shot. Even if the lab found relevant SNPs, it was likely few people would share the same polymorphisms, making drug development and testing nearly -- if not completely -- impossible.
Then they got lucky. The search for SNPs in the genetic material of 109 Huntington patients uncovered a single SNP carried by 48 percent of people with Huntington's. "This SNP is actually associated with the disease. We don't know why," Zamore said. That meant a single siRNA could shut off expression in the mutant Huntingtin gene - while leaving cells' healthy Huntingtin genes intact -- in almost half of all U.S. and European Huntington's patients.
"The most exciting part of the study was finding one siRNA that clearly is the top candidate for a clinical trial, where the patient population is predicted to be sufficiently large that it merits the development of a drug you could take into trial," Zamore said.
"That takes away the biggest worry we had, which was that the number of siRNAs we would have to test in order to have impact on the disease would be too large, and as a consequence the FDA would never approve any trial," he continued.
By adding an siRNA against one of two other common SNPs, Zamore says gene silencing could be effective in 75 percent of patients with Huntington's disease in the U.S. and Europe. Although the group found other SNPs, targeting more of them failed to increase the number of patients who could be helped, he said.
The next problem became developing siRNAs that could discriminate between target mRNAs and non-target. "That turned out to be frustratingly difficult," Zamore said. In tests of human cells, the siRNA sometimes sliced up the disease RNA, as it should. But sometimes it destroyed the normal Huntingtin RNA as well. To prevent this error, Zamore and his colleagues changed one more nucleotide base on the siRNA. Now, the silencing RNA was different from the healthy mRNA by two nucleotides, making it less likely to grab the good RNA.
Further research in mice will examine the efficacy of the siRNA tool. "The siRNA has to be sufficiently stable, and has to get into the right cells, and has to discriminate between the two (genes). It's incredibly expensive work," he said.
Zamore acknowledges that even with this progress, they're a long way from a treatment for Huntington's. "The Huntington's community is very savvy about understanding that scientific progress is always plodding. It's the sum of lots of little steps. From our perspective, the most important thing is to keep taking those steps."
Source: Howard Hughes Medical Institute (news : web)