Neuroscientists unveil molecular pathway involved with Huntington's disease
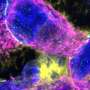
 and Htt (green) shows abundant expression of CalDAG-GEFI in the striatum of control mice (left panel) but severely down-regulated expression in striatal neurons of the Huntington's disease model mice (right panel). Aggregated Htt, a hallmark of Huntington's disease, is apparent in the neurons of mutant mice (green particles, right panel). Image courtesy Jill Crittenden/MIT")
(PhysOrg.com) -- MIT researchers have discovered new molecular changes in the brains of individuals with Huntington’s disease, a genetic disorder that leads to neuronal loss accompanied by unwanted movements, psychiatric symptoms, and eventual death. By studying brains of human patients, as well as mouse and rat models, they have uncovered a protective response that may eventually lead to new therapies for this currently incurable disease.
Huntington’s disease occurs in patients who inherit a mutant form of a protein called Huntingtin (Htt). The protein was first identified in 1993, but how it leads to disease is still poorly understood. One paradox is that the Htt protein is present throughout the body, yet the damage it causes is largely concentrated within specific populations of neurons in the striatum — a brain region also implicated in Parkinson’s disease and other disorders.
The MIT team led by Ann Graybiel, an Institute Professor and member of the McGovern Institute for Brain Research, focused on a gene known as CalDAG-GEFI, which is particularly enriched in the striatal neurons that die in Huntington’s disease. The MIT team showed that CalDAG-GEFI is dramatically down-regulated in the brains of individuals with Huntington’s disease as well as in mouse models of the disease. By following mutant mice for up to 9 months, the researchers showed that this reduction occurs gradually, in parallel with the progression of the disease.
These progressive changes suggest that CalDAG-GEFI is likely to play some role in the disease process. The researchers wanted to determine whether the suppression of this gene is part of the death process, or whether it represents part of the brain’s protective response. They found that the latter explanation appears to be true - when the researchers artificially blocked the expression of CalDAG-GEFI (using a method known as siRNA), the striatal neurons were protected from Htt-induced damage.
“So the enriched expression of CalDAG-GEFI in the striatum may explain, in part, why striatal neurons are particularly vulnerable to the expression of mutant Htt,” explained first author and research scientist, Jill Crittenden of the McGovern Institute for Brain Research. “Switching off of the CalDAG-GEFI gene may represent the neuron’s attempt, ultimately unsuccessful, to save itself.”
Huntington’s disease is currently incurable, and existing treatments address only the symptoms, and have no effect on the course of the disease or its eventual fatal outcome. The researchers hope that by understanding the molecular pathway by which neurons are killed, their findings may suggest new strategies for the development of treatments that could slow or even prevent the progression of the disease.
More information: Crittenden J, Dunn DE, Merali FI, Woodman B, Yim M, Borkowska AE, Frosch MP, Bates GP, Housman DE, Lo DC, Graybiel AM. CalDAG-GEFI Down-regulation in the striatum as a neuroprotective change in Huntington’s Disease. Human Molecular Genetics. 10 February 2010.