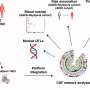
Disease-causing mutation disrupts movement of cell's 'power house'
 compared with how little mitochondria with mutated proteins travel (bottom graph). Credit: Courtesy, with permission: Misko et al. The Journal of Neuroscience 2010.")
New research shows how a mutation causes a common inherited neurodegenerative disease, according to a study in the March 24 issue of The Journal of Neuroscience. The study shows that the mutation of a specific protein known to cause Charcot-Marie-Tooth (CMT) disrupts the movement of mitochondria, the energy-supplying machines inside each cell. The regulated movement of mitochondria along nerve cell fibers is vital to normal communication between the brain and muscles.
A mutation in the protein mitofusin 2 had been known to cause one type of CMT, a common inherited neurological disorder characterized by a loss of muscle tissue and sensation in the limbs, which affects about 2.6 million people. In this study, a team of researchers lead by Robert Baloh, MD, PhD, of Washington University School of Medicine, examined the role of mitofusin proteins in the cell to learn more precisely how the mutation causes the disease.
"Our study provides the first evidence that mitofusins directly regulate the movement of mitochondria in nerve fibers," Baloh said. "Furthermore, our work suggests the basis for this particular form of CMT is the abnormal movement of mitochondria in these fibers."
Mitochondria are dynamic cellular power providers that travel to places in the cell where energy is needed. All this activity hinges on a series of molecular signals that regulate where mitochondria go.
Baloh and his colleagues used images of live cells taken from mice to study the movement of mitochondria, which moved slower in cells with mutated mitofusin 2, suggesting that the protein directly affects their transport. Until now, researchers had been unsure as to whether the abnormality lay in their transport along, or attachment to, nerve fibers.
"This discovery places this type of CMT in the ever-growing list of neurodegenerative diseases caused by transport problems and strengthens the possibility of using general enhancers of this process as therapy for different types of diseases," said Vincent Timmerman, PhD, of the University of Antwerp in Belgium, who was unaffiliated with the study.
The authors also suggest that a related protein called mitofusin 1 might someday serve to compensate for a mutated and malfunctioning mitofusin 2. Although mitofusins 1 and 2 are different proteins, they play similar roles in a cell. Baloh and his team suggest that mitofusin 1 may be able to perform the function of mitofusin 2 and regulate the transport of mitochondria. Finding a way to increase the levels of mitofusin 1 might have therapeutic effects for patients who have mutated mitofusin 2.