Over-abundant protein prompts neurodegenerative cascade
In diverse neurodegenerative diseases ranging from Parkinson's to Alzheimer's, researchers have long noted accumulations of a little-understood neuronal protein called α-synuclein. Pathological and genetic evidence strongly suggested that excessive α-synuclein played a role in the evolution of these diseases, but it was unclear how too much α-synuclein culminated in synaptic damage and neurodegeneration.
In a paper published in the June 16 issue of The Journal of Neuroscience, neuroscientists at the University of California, San Diego School of Medicine have taken steps toward elucidating the early deleterious impact of even modest over-accumulations of α-synuclein, describing a cascade of abnormal intracellular events that results in a phenomenon they call "vacant synapses," reduced transmissions among affected neurons, synaptic loss, and ultimately, dementia.
"One of the fundamental questions in neurodegeneration research is what are the early changes that make a brain go bad," said lead author and principal investigator Subhojit Roy, MD, PhD, a neuroscientist and neuropathologist at the department of neurosciences at UC San Diego School of Medicine and the Shiley Marcos Alzheimer's Disease Research Center.
"All a neuron really does is communicate. Extensive research has shown that deficiencies and defects in the act of communicating with other neurons are what cause neurodegeneration," said Roy. "While it's clear that even modest elevations of α-synuclein in neurons is pathogenic and that they impact neuronal communication, it is unclear how α-synuclein does it. If we can understand this process, then maybe interventional targets or therapies can be developed at early stages when these diseases would still be amenable to treatments."
Roy, with colleagues David A. Scott , Yong Tang, Anna Cartier and Eliezer Masliah, all in the UCSD department of neurosciences, and Iustin Tabarean at The Scripps Research Institute, developed a model-system in which they could study thousands of neurons modestly over-expressing α-synuclein.
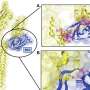
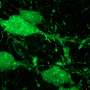
The researchers cultured neurons from a transgenic mouse brain in which α-synuclein molecules were tagged with a green fluorescent protein. As α-synuclein levels accumulated in the cells, the neurons would get greener and greener, "leading to a sea of α-synuclein-positive neurons, an experimentalist's delight!" said Roy. Meanwhile, levels of other synaptic proteins critical to neuronal communication diminished and disappeared. These changes or vacant synapses correlated with severe defects in neurotransmitter release.
Simply put, in the presence of excessive α-synuclein, the affected neurons stopped doing what neurons do - communicate.
"Based on our findings, we propose a new disease model where excessive α-synuclein triggers a pathologic chain of early events that eventually lead to the loss of critical synaptic proteins and decreases in neurotransmitter release, causing synaptic dysfunction and - ultimately - dementia," said Roy. Related neuropathologic examinations of brain samples of human patients also support this overall idea.
This pathologic cascade of neurodegeneration induced by α-synuclein has not been shown before, but Roy said many details and questions remain to be addressed. For example, what causes the α-synuclein imbalance and how does the protein reduce the levels of other proteins?
One possible answer to the latter question, said Roy, is that aggregations of α-synuclein act like a physical barrier, preventing other proteins from reaching synapses from the neuron's main body. "Like dumping rocks in a road, they become an obstacle to things trying to get to past them," Roy said. "The idea that α-synuclein blocks transport is well-supported by previous studies in yeast and other reduced systems. We have no favorite hypothesis - only a model-system where we can see things unfold with exceptional clarity. We will continue to look and see what our green neurons tell us."