Study implicates new epigenetic player in mental retardation and facial birth defects

A subtle mutation affecting the epigenome -- a set of dynamic factors that influence gene activity -- may lead to an inherited form of mental retardation that affects boys, find researchers at Children's Hospital Boston. The disorder, which also involves cleft lip or cleft palate, appears to hinge on an enzyme working in a biological pathway that may offer several potential drug targets.
The study, published online July 11 in the journal Nature, reveals that this enzyme is a histone demethylase and works with a key genetic partner to help keep neuronal cells alive during development of the embryonic brain. Patients with this form of mental retardation are known to have mutations in the gene that encodes the active part of this enzyme. The findings may help scientists further understand the underlying biological reasons why X-linked disorders cause cognitive impairment and develop new therapies to treat or prevent them.
"Human genetics has made great strides in identifying genes as potential causes of diseases and disorders, but we don't know much about how they work," says senior author Yang Shi, PhD, the Merton Bernfield Professor of Neonatology in the Newborn Medicine division at Children's. "We knew this was a biologically relevant gene. We wanted to understand the etiology, so we asked why the gene causes problems when it is mutated. Here, we have identified a direct target in neuronal and craniofacial development."
The fast-moving young field known as epigenetics is revealing the dynamic structures and processes that organize, index and control access to the information stored in the DNA code. The epigenetic program orchestrates different combinations of gene activity - allowing cells with identical genomes to be transformed into more than 200 different specialized tissues and organs in our bodies.
When most people think of DNA, they picture the iconic spiraling ladder of naked DNA. But in nature, the twisting double-helix strands actually spool around clusters of proteins called histones with protruding "tails" that act like specialized antennas, transmitting directions for DNA. This dynamic structure, called chromatin, extends the genetic code by offering, measuring or limiting access to different genes.
Several years ago, Shi and his colleagues identified the first enzyme that can detach a molecule known as a methyl group, previously thought to be a permanent fixture, from the histones tails. Then his team and a number of other research groups independently discovered members of a second known family of these enzymes, known collectively as histone demethylases.
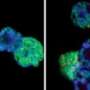
The latest study began with a gene mutated in several male patients with X-linked mental retardation and craniofacial abnormalities. The gene codes for an enzyme that looked a lot like a member of the second family of histone demethylases. The mutations in these patients abolished the working part of the enzyme that plucks the methyl group from the histone tail.
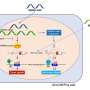
Led by Hank Qi, PhD, co-first author and postdoctoral fellow, the researchers demonstrated in human cells that the enzyme, PHF8, indeed works as a histone demethylase. (And it is the first known demethylase discovered for a strategic methylation point on the tail of histone 4 known as H4K20, which other evidence suggests plays a critical role in gene expression and regulation and in the DNA damage response.) In this case, by removing the methyl group, the enzyme appears to maintain active gene transcription.
"The histone methylation and demethylation doesn't turn the gene on or off," Qi says. "When this histone mark changes, it generates an equilibrium important for fine-tuning gene expression."
Despite its widespread presence, the enzyme seems to have a narrowly targeted biological effect on a master genetic regulator of craniofacial development, the transcription factor MSX1. Taking a cue from the scientific literature, Qi and his collaborators, Madathia Sarkissian and Thomas Roberts at Dana-Farber Cancer Institute, tested the normal enzyme function in zebrafish, a popular model for genetic function.
It is hard to judge cognitive impairment in a small fish, but the dramatic impact on craniofacial development was obvious. Fish without the enzyme developed virtually no jawbone, a condition that could be prevented by providing the functioning enzyme, showing its importance in development. As importantly, providing more of the fish version of the MSX1 gene (whose activity the demethylase enzyme encourages) also partially prevented the biological defects caused by the missing enzyme.
Hope for the reversibility of some aspects of mental retardation arose three years ago in a Scottish mouse study of Rett syndrome, a disorder on the autism spectrum that is also a cause of severe mental retardation in girls. The disease is caused by a molecule, MeCP2, that binds to methylated DNA and may be involved in another form of epigenetic regulation.
"In practical terms, we use gene expression as a read out," says Shi, also a professor of pathology at Harvard Medical School. "Epigenetic states affect the expression of critical genes. These studies suggest that the imbalance of histone methylation dynamics plays a critical role in mental retardation. You can imagine a therapeutic approach to enhance the compromised enzymatic activity or to restore the downstream function."