Why do neurons die in Parkinson's disease?
Current thinking about Parkinson's disease is that it's a disorder of mitochondria, the energy-producing organelles inside cells, causing neurons in the brain's substantia nigra to die or become impaired. A study from Children's Hospital Boston now shows that genetic mutations causing a hereditary form of Parkinson's disease cause mitochondria to run amok inside the cell, leaving the cell without a brake to stop them. Findings appear in the November 11 issue of Cell.
Mitochondrial movement is often a good thing, especially in neurons, which need to get mitochondria to cells' periphery in order to fuel the axons and dendrites that send and receive signals. However, arresting this movement is equally important, says senior investigator Thomas Schwarz, PhD, of Children's F.M. Kirby Neurobiology Center, since it allows mitochondria to be quarantined and destroyed when they go bad.
"Mitochondria, when damaged, produce reactive oxygen species that are highly destructive, and can fuse with healthy mitochondria and contaminate them, too," Schwarz says. "It's the equivalent of an environmental disaster in the cell."
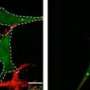
Studying neurons from fruit flies, rats and mice, as well as cultured human cells, Schwarz and colleagues provide the most detailed understanding to date of the effects of the gene mutations, which encode the proteins Parkin and PINK1. They demonstrate how these proteins interact with proteins responsible for mitochondrial movement -- in particular Miro, which literally hitches a molecular motor onto the organelle.
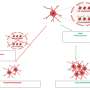
Normally, when mitochondria go bad, PINK1 tags Miro to be destroyed by Parkin and enzymes in the cell, the researchers showed. When Miro is destroyed, the motor detaches from the mitochondrion. The organelle, unable to move, can then be disposed of: The cell literally digests it.
But when either PINK1 or Parkin is mutated, this containment system fails, leaving the damaged mitochondria free to move about the cell, spewing toxic compounds and fusing to otherwise healthy mitochondria and introducing damaged components.
The study's findings are consistent with observed changes in mitochondrial distribution, transport and dynamics in other neurodegenerative diseases such as Huntington's disease, Alzheimer's disease, amyotrophic lateral sclerosis (Lou Gehrig's disease), and Charcot-Marie-Tooth disease, the researchers note.
Although the team studied a rare hereditary form of Parkinson's, the findings may shed light on what's going on in the more common sporadic form of the disease, Schwarz says.
"Whether it's clearing out damaged mitochondria, or preventing mitochondrial damage, the common thread is that there's too much damage in mitochondria in a particular brain region," he says.
While Schwarz sees potential in gene therapy to restore normal PINK1 or Parkin to neurons, he is more interested in the possibility of helping neurons flush out bad mitochondria or make enough new, healthy mitochondria to keep them viable. "We may need to do both," he says.