Gene therapy research cures retinitis pigmentosa in dogs
Members of a University of Pennsylvania research team have shown that they can prevent, or even reverse, a blinding retinal disease, X-linked Retinitis Pigmentosa, or XLRP, in dogs.
The disease in humans and dogs is caused by defects in the RPGR gene and results in early, severe and progressive vision loss. It is one of the most common inherited forms of retinal degeneration in man.
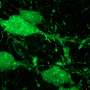
"Every single abnormal feature that defines the disease in the dogs was corrected following treatment," said lead author William Beltran, assistant professor of ophthalmology at Penn's School of Veterinary Medicine.
"We were thrilled," said senior author Gustavo Aguirre, professor of medical genetics and ophthalmology at Penn Vet. "The treated cells were completely normal, and this effect resulted from introducing the normal version of the human gene into the diseased photoreceptor cells."
The similarities between humans and dogs, in terms of both eye anatomy, physiology, disease characteristics and positive response to this gene therapy, raise hope for a clear path to human therapies.
Beltran and Aguirre collaborated with Artur Cideciyan and Samuel Jacobson at the Scheie Eye Institute, part of the University of Pennsylvania's Perelman School of Medicine. This achievement results from more than 10 years of close collaboration between the scientists at Penn's veterinary and medical Schools and the University of Florida.
In addition to others at Penn Vet, Scheie and Florida, researchers at the universities of Michigan and Massachusetts and the National Eye Institute at the National Institutes of Health contributed to the research.
The study will be published in the journal Proceedings of the National Academy of Sciences.
The gene therapy approach used takes advantage of a viral vector — a genetically modified virus that doesn't cause disease and is unable to divide -- to deliver the therapeutic RPGR gene specifically to diseased rods and cones. In the absence of treatment, these cells malfunction and progressively die.
The research team has previously successfully applied a similar approach to two other heritable vision disorders that occur in both humans and dogs: Leber congenital amaurosis and achromatopsia. The present study was more challenging, as it was necessary to target both main classes of photoreceptor cells.
While the exact disease mechanism of the RPGR form of XLRP is still unknown, the researchers were able to successfully treat dogs with two different RPGR mutations. The mutations disrupt photoreceptors in different ways, but both ultimately cause them to become useless for vision. While this form of blindness is rare in dogs, it is common in humans. Patients with XLRP usually begin to lose night vision as children and become almost totally blind by middle age.
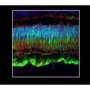
This is the first proof that this condition is treatable in an animal model; a single subretinal injection administered to the diseased dogs led to functional and structural recovery. The dogs' recovery was assessed using a variety of methods that are used clinically in patients, such as electroretinography and optical coherence tomography.
The researchers feel the results are promising and relevant for translation to the clinic.
"We are intervening to treat both classes of photoreceptor cells, rods and cones, and that has never been done before in a large animal model," Beltran said. "And not only can we prevent the disease onset but also restore the remaining photoreceptors cells to normal once the disease is ongoing."
While the ability to repair both rods and cones was itself a first, the research team went further, showing that its treatment also repaired the photoreceptor connections to other retinal neurons that eventually send visual signals to the brain, another first.
"This not only provides hope for reversing XLRP but potentially for any form of photoreceptor degeneration," Aguirre said. "Altered inner retinal wiring is a common feature for these diseases that has been considered irreversible.
"The study required a combination of genetic tools and surgical technique to make sure the therapy targeted only the diseased cells. The viral vector had to be injected in the sub-retinal space so as to be in close proximity to the photoreceptors. Likewise, you need to ultimately deliver the therapy to the right location of the retina," Aguirre said.
"In the human disease, careful characterization of the areas of the retina that need to be treated is going to be critical for therapy to succeed in the clinic," Cideciyan said.
The genetic aspect of the viral vector used in this study involved a double safeguard. The first safety feature was to use a viral vector that is known to predominantly target both rods and cones but not other cells. The second safeguard involved attaching the healthy RPGR gene to a "promoter," a piece of genetic code that would "switch on" the gene only if the virus penetrated the correct cell.
Selecting the right promoter was critical; the lead researchers at the University of Florida, William W. Hauswirth and Alfred S. Lewin, had to find one that that would be turned on exclusively in rods and cones. This way, even if the virus made its way to a non-photoreceptor cell, that cell would not start activating the RPGR gene.
That both the promoter and the RPGR gene it activates are taken from humans is a strong sign that the treatment may be translatable to patients.
"While there is still much work to do to assess long-term efficiency and safety with this approach, there is hope that this vector and knowledge could be used in a few years to treat the many patients losing vision from XLRP," Jacobson said.