Link between inflammatory process and progression of Alzheimer's disease
An international team of researchers from the University of Massachusetts Medical School, the University of Bonn and the Center for Advanced European Studies and Research in Germany have shown that a well-known immune and inflammatory process plays an important role in the pathology of Alzheimer's disease. This process, which results in the mature production of the pro-inflammatory cytokine called interleukin-1 beta (IL-1B) and is involved in the body's defense against infection, has also been established as a clinical target for rheumatoid arthritis. The finding, published in Nature, points to the possibility that drugs that disrupt the production of IL-1B, such as those for rheumatoid arthritis, may also prove beneficial for patients with Alzheimer's.
"This finding represents an important new clinical target for patients with Alzheimer's disease," said Douglas T. Golenbock, MD, chief of infectious diseases and immunology and professor of medicine and microbiology & physiological systems. "We've known for years that the plaques associated with Alzheimer's were surrounded by microglia, the resident immune cell of the central nervous system. What we didn't know was what role, if any, inflammation played in the progression of the disease. With this link we have a new path to potentially identifying and attacking this horrible disease."
The most common form of dementia, Alzheimer's is a degenerative neurological disorder that leads to memory loss, impaired cognitive function, and eventually death. By 2050 it is predicted that 1 in 85 people will suffer from Alzheimer's disease. There are no available treatments.
A key physiological component of Alzheimer's disease is the presence of extracellular plaques, primarily composed of beta amyloid peptides, which aggregate in the brain. These plaques are believed to be toxic and the chief cause of nearby neuron death and cortical material loss. The hippocampus, which plays an important role in short-term memory, is one of the first regions of the brain to suffer damage from Alzheimer's.
Golenbock and colleagues had established in previous studies that neurons in cell cultures died after nearby microglia cells, the main form of active immune defense in the brain and spinal cord, were exposed to amyloid beta fibrils, such as those found in Alzheimer's plaques. Typically, microglias are responsible for removing plaques, damaged neurons and infectious agents from the central nervous system. The beta amyloid peptide, however, generates inflammation in the central nervous system by activating microglia to produce neurotoxic compounds, including cytokines. How this process was being activated in patients with Alzheimer's disease, though, was unclear.
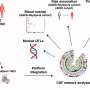
Earlier work done in the Golenbock laboratory demonstrated that beta amyloid peptide could induce the production of IL-1B by activating a multi-protein receptor complex in microglial cells known as the NLRP3 inflammasome. Because of its ability to sense the beta amyloid peptide, the NLRP3 inflammasome has been implicated in several chronic inflammatory diseases, including gout and asbestosis. Examining Alzheimer's tissue samples, scientists found that "every one of the cell samples contained increased evidence of activated inflammasomes, strongly suggesting that they were producing IL-1B," said Golenbock.
"Taken together with our earlier studies, this strongly suggested a role for NLRP3 and caspase-1 in producing IL-1B leading to Alzheimer's disease progression," Golenbock said.
To assess the precise impact of NLRP3 and caspase-1 on Alzheimer's disease in an organism, researchers recorded cognitive function and memory in mice models that expressed genes associated with familial Alzheimer's but that were deficient in NLRP3 or caspase-1, and compared them with Alzheimer's mice that had otherwise intact immune systems. When researchers performed memory recall tests of the Alzheimer's in NLRP3- or caspase-1- mutant mice, they found the animals exhibited far better memory recall and appeared protected from memory loss. However, Alzheimer's mice that expressed NLRP3 and caspase-1 at normal levels exhibited symptoms consistent with Alzheimer's disease. Further examination revealed that NLRP3 and caspase-1 deficient mice showed a decrease in beta amyloid plaques and in increased ability of the microglia to remove fibrillar beta amyloid from the brain.
It was also revealed that activated IL-1 levels in the NLRP3 and caspase-1deficient mice were reduced compared to symptom-bearing counterparts. Because of NLRP3 and caspase-1 deficits, these mice produced less IL-1. These deficits appeared to promote formation of a microglia cell phenotype which was more capable of metabolizing and removing Alzheimer's plaques from the central nervous system.
"These findings suggest that a knockout of NLRP3, caspase-1 or mature IL-1B may represent a novel therapeutic intervention for Alzheimer's disease," said Golenbock. "It's possible that drugs that block NLRP3 or IL-1B – including some of which are already in clinical trials or on the market – might provide some benefit," said Golenbock.
"The critical part, though, is how much NLRP3 or IL-1B production can these drugs disrupt," said Golenbock. "I believe that it's not enough to block just 90 percent; it will probably have to be closer to 100 percent."
More information: NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice, Nature, DOI: 10.1038/nature11729