Silencing sudden death: Study targets genetics of hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy (HCM), a disease in which cardiac muscle thickens, weakening the heart, can be prevented from developing for several months in mice by reducing production of a mutant protein, according to a new study by researchers at Harvard Medical School.
The work takes a first step toward being able to treat or prevent the leading cause of sudden death in athletes and sudden heart-related death inpeople under 30 in the United States.
"There's really no treatment for HCM right now. You can treat symptoms like chest pain or an arrhythmia, but that's not getting at the fundamental problem," said Christine Seidman, the Thomas W. Smith Professor of Medicine and Genetics at HMS and Brigham and Women's Hospital, a Howard Hughes Medical Investigator and senior author of the study. "While the application of this strategy is in the very early stages, it shows considerable promise."
The results were published in Science on Oct. 3.
An estimated 1 in 500 Americans has HCM. Although many of them never develop symptoms, for others the disease can be severe or fatal.
More than 1,000 different mutations that can cause HCM have been identified across about 10 genes that make heart muscle proteins. People with HCM have one "good" copy and one "bad" copy of one of those genes.
Studying one of the mutations that causes particularly severe disease, Christine Seidman and Jonathan Seidman, Henrietta B. and Frederick H. Bugher Foundation Professor of Genetics at HMS, worked with research fellow Jianming Jiang and instructor Hiroko Wakimoto to target the analogous "bad" gene in mice while leaving the "good" gene alone.
The researchers created an RNA interference (RNAi) tool designed to home in on the single HCM-causing mutation and stop it from making its harmful protein. They packaged the RNAi inside a virus (a common RNAi delivery technique) and injected it into lab mice engineered to develop HCM. They compared the results to two untreated groups of mice: one with the same HCM mutation, and one without.
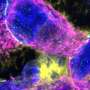
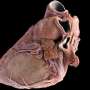
By suppressing the "bad" gene, the RNAi was able to reduceproduction of the mutant protein by about 28 percent. That was enough to prevent development of HCM manifestations—including ventricular wall overgrowth, cell disorganization and fibrosis (scarring)—for about six months, or one-quarter of the mice's lifespans.
"For all intents and purposes, the heart looked normal," said Christine Seidman. "Wonderfully, boringly normal."
The treatment successfully targeted heart cells in the mice without affecting other organs. Although it did not reverse any existing HCM damage, Jonathan Seidman noted that halting the progress of HCM would be a significant advance in itself.
"If somebody already had a certain amount of wall thickness and you prevent it from worsening, that would be a step forward to limit progressive symptoms and development of heart failure," he said.
In addition to its potential for informing HCM treatment in humans down the road, the initial findings could be relevant for a related genetic condition called dilated cardiomyopathy, where the heart becomes baggy and thin-walled and contracts too little instead of too much.
The researchers now plan to investigate whether they can continue to delay HCM in mice with booster shots, reverse disease damage or reduce HCM-related arrhythmias. They would like to study a larger animal model as well as explore whether younger mice respond better to therapy than older mice and if interventions aimed at specific areas of the heart could be as effective as treating the whole heart.
The team also intends to explore whether a collection of 10 RNA is could be engineered to target HCM genes instead of having to develop 1,000 RNAis to target individual mutations.
More information: "Allele-Specific Silencing of Mutant Myh6 Transcripts in Mice Suppresses Hypertrophic Cardiomyopathy," by J. Jiang et al. Science, 2013.