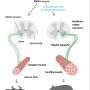
Researchers track down cause of eye mobility disorder
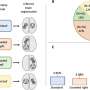
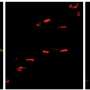
 as compared to an incomplete nerve, a condition resulting in permanent downward gaze in both mice and humans. Credit: Jeremy Duncan.")
Imagine you cannot move your eyes up, and you cannot lift your upper eyelid. You walk through life with your head tilted upward so that your eyes look straight when they are rolled down in the eye socket. Obviously, such a condition should be corrected to allow people a normal position of their head. In order to correct this condition, one would need to understand why this happens.
In a paper published in the April 16 print issue of the journal Neuron, University of Iowa researchers Bernd Fritzsch and Jeremy Duncan and their colleagues at Harvard Medical School, along with investigator and corresponding author Elizabeth Engle, describe how their studies on mutated mice mimic human mutations.
It all started when Engle, a researcher at the Howard Hughes Medical Institute (HHMI), and Fritzsch, professor and departmental executive officer in the UI College of Liberal Arts and Sciences Department of Biology, began their interaction on the stimulation of eye muscles by their nerves, or "innervation," around 20 years ago.
Approximately 10 years ago, Engle had identified the mutated genes in several patients with the eye movement disorder and subsequently developed a mouse with the same mutation she had identified in humans. However, while the effect on eye muscle innervation was comparable, there still was no clue as to why this should happen.
Fritzsch and his former biology doctoral student, Jeremy Duncan, worked with the Harvard researchers on a developmental study to find the point at which normal development of eye muscle innervations departs from the mutants. To their surprise, it happened very early in development. In fact, they found—only in mutant mice—a unique swelling in one of the nerves to the eye muscle.
More detailed analysis showed that these swellings came about because fibers extending to the eyes from the brain tried to leave the nerve as if they were already in the orbit, or eye socket. Since it happened so early, the researchers reasoned that something must be transported more effectively by this mutation to the motor neurons trying to reach the orbit and the eye muscles; something must be causing these motor neurons to assume they have already reached their target, the orbit of the eye.
To verify this enhanced function, the researchers developed another mouse that lacked the specific protein and found no defects in muscle innervation. Moreover, when they bred mice that carried malformed proteins with those that had none of these proteins, the mice developed a normal innervation.
This data provided clear evidence of what was going wrong and why, but it did not provide a clue as to the possible product that was more effectively transported in the mutant mice and, by logical extension, in humans. Further analysis revealed that breeding their mutant mice with another mutant having eye muscle innervation defects could enhance the effect of either mutation.
With this finding, they had identified the mutated protein, its enhanced function, and at least some of the likely cargo transported by this protein to allow normal innervation of eye muscles. This data provides the necessary level of understanding to design rational approaches to block the defect from developing.
Knowing what goes wrong and at what time during development can allow the problem to be corrected before it develops through proper manipulations. Engle, Fritzsch, and their collaborators currently are designing new approaches to rescue normal innervation in mice. In the future, their work may help families carrying such genetic mutations to have children with normal eye movement.