Can anti-depressants help prevent Alzheimer's disease?
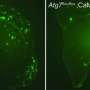
 dot the brain of a mouse model of Alzheimer's disease. Scientists have found that an antidepressant can reduce production of the primary component in these plaques. Credit: John Cirrito, Ph.D.")
A University of Pennsylvania researcher has discovered that the common selective serotonin reuptake inhibitor (SSRI) citalopram arrested the growth of amyloid beta, a peptide in the brain that clusters in plaques that are thought to trigger the development of Alzheimer's disease (AD). Penn, in collaboration with investigators at Washington University, tested the drug's effects on the brain interstitial fluid (ISF) in plaque-bearing mice and the cerebrospinal fluid (CSF) of healthy human subjects to draw its conclusions, which are detailed in the new issue of Science Translational Medicine.
Alzheimer's disease is the sixth leading cause of death the United States, affecting five million patients, with the numbers expected to leap to approximately 16 million patients in the coming decades, unless preventive measures are developed.
"Our previous studies have shown an association between anti-depressants and the reduction in amyloid burden in the brain," says the paper's lead author, Yvette Sheline, MD, professor of Psychiatry, Radiology and Neurology and director of the Center for Neuromodulation in Depression and Stress, at Penn's Perelman School of Medicine. "Those studies examined a retrospective correlation between the duration of anti-depressant use and amyloid burden shown in PET scans in the brains of elderly volunteers. With this new study we took our research a step further and tested the prospective effect of the SSRI citalopram on the CSF amyloid levels in younger, healthy subjects." Sheline performed the research while at Washington University.
She again found that citalopram, which is approved by the FDA in 1998 for the treatment of depression, had significant effects.
The brain interstitial fluid (ISF) of transgenic plaque-bearing mice following exposure to citalopram showed that the level of amyloid-beta in the ISF decreased in a dose-dependent manner by as much as 25 percent compared to baseline numbers. In addition, the researchers found that two months of citalopram exposure in plaque-bearing mice resulted in no new plaque development, and no growth of existing plaques compared with a marked increase in plaque growth and development in the control group of mice, who were exposed to sugar water. However, citalopram had little effect on the regression of already existing amyloid plaques.
In a parallel study, 23 healthy human subjects, age 18 to 50 without medical disease and with no previous history of anti-depressant treatment, were administered 60 mg citalopram, roughly equivalent to the dose used in mice.
The double-blind study showed that citalopram was associated with a 38 percent lower A-beta concentration over the 37-hour testing period versus placebo, and showed a reduction in newly-produced A-beta in the citalopram-treated group versus the control group.
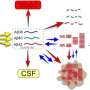
SSRIs are thought to produce their antidepressant effect by blocking the reuptake of the neurotransmitter serotonin into the presynaptic terminals of the neurons, increasing the availability of serotonin and reducing A-beta production. Serotonin receptor levels are reduced in brains of patients with AD. In contrast, this newly described effect on the reduction of amyloid protein concentration most likely occurs by a different pathway.
The development of safe and effective therapeutic approaches that can reduce CSF A-beta production even modestly may prevent a cascade of neuronal damage, which would have an important impact on preventing or slowing progression to symptomatic AD.
"While these results are an excellent start at lowering A-beta production, we are a long way from making a statement regarding the ability of SSRIs to prevent the cognitive decline associated with AD," Sheline says. "We are developing a greater understanding of the capabilities of SSRIs, which offer promise for the future as preventive measures, as we continue to uncover the complex mechanisms in the brain that trigger Alzheimer's and dementia."
More information: "An Antidepressant Decreases CSF Aβ Production in Healthy Individuals and in Transgenic AD Mice," by Y.I. Sheline et al. Science Translational Medicine, 2014.