Study shows how misfolded proteins are selected for disposal

It's almost axiomatic that misfolded proteins compromise how cells normally function and cause debilitating human disease, but how these proteins are detected and degraded within the body is not well understood. Neurodegenerative diseases – including Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis (Lou Gehrig's disease), Huntington's disease, and spinocerebellar ataxias – exact a devastating toll on aging populations throughout the world.
"Yet, there is virtually no cure for any of these diseases, and clinical trials have yielded mostly disappointing results, indicating that investigators are missing something fundamental about these diseases," says Xiaolu Yang, PhD, professor of Cancer Biology, Perelman School of Medicine at the University of Pennsylvania. All of these diseases are caused by the accumulation of toxic misfolded proteins in different types of neurons. "However, our knowledge about how cells normally remove these proteins, an issue fundamental to understanding what goes wrong in neurodegenerative diseases, is very limited."
Yang and first author Lili Guo, PhD, who was a doctoral student in the Yang lab, identified a protein recycling pathway in mammalian cells that removes misfolded proteins. Their findings appear online today in Molecular Cell ahead of the print edition. They also demonstrated this pathway's role in protecting against neurodegenerative diseases in an animal model. Guo is now a postdoctoral fellow in another Perelman School lab.
Proteins are the work horses of the cells. They are the most abundant macromolecules, extremely versatile in their functions and critically important for virtually all biological processes. However, proteins are also highly prone to misfolding due to genetic mutations, synthetic inaccuracies, and irreparable damages. What's more, the half-life of many proteins is relatively short, from a few minutes to a few hours, necessitating their continued synthesis. As such this puts a burden on the cell to maintain quality control in the correct folding of proteins.
"This paper resolves a long-standing question in protein quality control of how misfolded proteins are precisely selected for degradation," notes Yang. "This newly described system could be a valuable target for treating neurodegenerative diseases."
Two-Staged Recycling
In any normally functioning cell, two systems maintain protein quality. First, chaperone proteins, like fingers that fold paper into origami shapes, guide amino acid chains in folding into their final, proper protein forms. Second, the recycling systems dispose of misfolded proteins and ultimately breaks them up into individual amino acids. This system involves the proteasome, a protein complex found throughout the cytoplasm and nucleus of cells. "But it is unclear how misshapen proteins are recognized and shuttled to the proteasome to be degraded. This study moves the field forward because we showed that the system is common for many types of misfolded proteins," notes Yang.
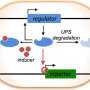
In addition to identifying the step-by-step molecular players of the system that eliminates misfolded proteins, they also defined the system's method of action. The mechanism of action is a relay system with two proteins.
The first protein, PML/TRIM19, recognizes features of misfolded proteins such as exposed water-phobic inner cores, which stick out and enhance the forming of toxic protein clumps. PML selectively interacts with misfolded proteins by recognizing this and other distinct features and tags the misfolded proteins with chains of a small protein called SUMO (small ubiquitin-like modifier). The SUMO-modified misfolded proteins are then recognized by the second protein, RNF4, which tags them with chains of another small protein called ubiquitin. The ubiquitin chain is a signal that can be recognized by the barrel-shaped proteasome, leading to the degradation of misfolded proteins.
The team then went on to demonstrate the physiological importance of the elimination system using a mouse model of spinocerebellar ataxia 1 (SCA1), a fatal neurological disorder that causes problems with movement and balance. Mutations in the ATXN1 gene cause SCA1, which involves repeated segments of the DNA building blocks cytosine (C), adenine (A), and guanine (G) that appear multiple times in a row in the gene, which encodes a tract of contiguous glutamine amino acids. Normally, the CAG segment is repeated 4 to 39 times within the gene. In SCA1, the CAG segment is repeated 40 to more than 80 times, which leads to an abnormally long ataxin-1 protein that folds into the wrong 3-dimensional shape and forms clumps within the nucleus. The team showed that a PML deficiency exacerbates both the behavioral and neuropathological defects caused by the expanded CAG repeat in the SCA1 mouse, indicating that PML normally operates to protect against neurodegeneration.
The PML protein is also involved in the formation of tumors, and that's how Yang, a cancer biologist, got involved in this type of research.
"The knowledge gained from this project now provides for the possibility of new therapeutic targets for neurodegenerative diseases. Perhaps new drugs could enhance the elimination system by increasing the action of PML or RNF4, or could inhibit the inhibitors of the SUMO and ubiquitin tagging process," says Yang